超高效液相色谱-串联质谱法检测猪肉中多兽药残留
2020-09-10杨兆甜吴亚婕王雅静吕青骎
杨兆甜, 吴亚婕, 王 莹, 王雅静, 吕青骎,2, 王 玮*,2
(1. 南京农业大学 食品科技学院, 江苏 南京210095;2. 南京农业大学 农业农村部肉及肉制品质量监督检验测试中心(南京),江苏 南京210095)
目前,我国在畜牧养殖方面使用较多的兽药有β-受体激动剂类、磺胺类、砜类抑制剂类以及喹诺酮类等[1-2]。 兽药的滥用易使畜牧产品遭受严重污染[3],药品经代谢进入动物源性食品中,之后可通过食物链在人体蓄积, 不仅能使人体产生抗药性、引起过敏反应、影响肠道内菌群[4],还能导致中毒等不良反应[5-6]。 因而,动物源性食品的兽药残留问题引起了广泛关注[7-9]。
目前,动物源性食品兽药残留的检测方法主要有毛细管电泳法 (Capillary Electrophoresis,CE)[10]、高效液相色谱法 (High Performance Liquid Chromatography,HPLC)[11-12]、免疫法(Immunoassay,IA)[13-14]和超高效液相色谱-串联质谱法 (Ultra Performance Liquid Chromatography/tandem Mass Spectrometry,UPLC-MS/MS)[15-16]等。 其中,CE 法可高效分离化合物,但重现效果不理想[17];HPLC 法检测快速、价格较低,但灵敏度较差[18-19];IA 法检测快速、灵敏,但假阳性率偏高[20]。 而且以上方法大多按照兽药种类分成若干种[21-22],检测效率较低,且兽药高通量同时检测分析受制于缺乏统一的前处理方法难以实现[23-24]。作者在采用Oasis PRiME HLB 固相萃取小柱净化和提取药物的基础上,通过优化前处理方法,建立了一种高通量快速测定猪肉中20种兽药残留的UPLC-MS/MS 方法。 该方法可以弥补常规检测方法的缺陷,旨在为我国动物源性食品安全监测体系建设、国际贸易中药物残留检测提供技术保障,同时有助于提高我国动物源食品的安全性,维护我国消费者的权益。
1 材料与方法
1.1 试验仪器与材料
advance EDI 纯水仪: 购于德国Sartorius 公司;I-class & Xevo TQ-S Micro型超高效液相色谱质谱仪: 购于美国Waters 公司;N-EvAP型24 管氮吹仪: 购于美国Organomation 公司;D-16C 高速冷冻离心机:购于德国Sartorius 公司;Oasis PRiME HLB小柱:购于美国Waters 公司。
1.2 试验试剂
试验材料为市售生鲜猪肉,购于江苏省南京市超市及农贸市场。
标准品:沙丁胺醇、莱克多巴胺、特布他林、克伦特罗、妥布特罗、氯丙那林、磺胺甲恶唑、磺胺甲氧嘧啶、磺胺二甲嘧啶、磺胺喹噁啉、氨苯砜、达氟沙星、沙拉沙星、恩诺沙星、环丙沙星、洛美沙星、培氟沙星、地塞米松、替米考星、红霉素、沙丁胺醇-D3、莱克多巴胺-D3、特布他林-D9、克伦特罗-D9、妥布特罗-D9、氯丙那林-D7、恩诺沙星-D5:购于德国Dr. Ehrenstorfer 公司;甲醇(色谱纯)、乙腈(色谱纯)、甲酸(色谱纯):购于美国Fisher 公司。
1.3 试验方法
1.3.1 样品的前处理准确称取生鲜猪肉样品制得的肉糜2.50 g,将其放置于50 mL 离心管中,依次加入20 μL 内标混合工作液和10 mL 体积分数80%的乙腈水溶液, 涡旋振荡1 min, 超声提取3 min,10000 r/min 离心5 min,获取上清液。 在固相萃取装置上放置Oasis PRiME HLB 小柱, 取5 mL上清液加载到固相萃取柱,收集过滤液。 并取4 mL滤液在50 ℃水浴的条件下用氮气吹干,经1 mL 体积分数为10%的甲醇水溶液复溶后用0.22 μm 微孔滤膜过滤,复溶液进行UPLC-MS/MS 分析。 采用阴性样品参照上述步骤进行空白试验;在阴性样品中加入混合标准溶液,并参照上述步骤进行加标试验。
1.3.2 标准溶液的配制单标储备液配制:准确称取20种待测药物的标准品,以甲醇溶液作为溶剂,配制为一定质量浓度(20~100 μg/mL)的单标储备液。
6 类药物混合标准溶液配制: 分别取同类别中一定量的单标储备液并用甲醇定容,使6 类药物混合标准溶液的质量浓度均为10.00 μg/mL。
混合标准溶液的配制:分别移取6 类药物混合标准溶液1.00 mL,并用甲醇溶液定容至10.00 mL,得到1.00 μg/mL 的混合标准溶液。
内标溶液的配制:各种内标以甲醇溶液为溶剂配制得质量浓度为10 μg/mL 的内标储备液; 分别移取各种内标储备液1.00 mL, 用甲醇溶液定容至10.00 mL,得到1.00 μg/mL 的内标混合溶液。
1.3.3 UPLC-MS/MS 法的建立色谱条件:BEH C18色谱柱(2.1 mm×50 mm,1.7 μm);以体积分数0.1%的甲酸水溶液和体积分数0.1%的甲酸乙腈溶液作为流动相;流速保持在0.3 mL/min;柱温保持在30℃;进样量为10 μL;色谱柱的洗脱方式采用梯度洗脱,其梯度洗脱条件如表1 所示。
质谱条件: 电喷雾正离子模式(electrospray ionization in positive mode,ESI+); 扫描模式为多反应监测(multiple reaction monitoring,MRM);脱溶剂气流量为1000 L/h;锥孔气流量为20 L/h;离子源温度保持在500 ℃。 监测参数见表2。

表1 梯度洗脱条件Table 1 Conditions of gradient elution
1.3.4 检出限、定量限以及线性范围的确定参照1.3.3 的色谱条件和质谱条件进行测定,以3 倍信噪比(S/N=3)计算检出限(limit of detection,LOD),10倍信噪比(S/N=10)计算定量限(limit of quantitation,LOQ)。 在猪肉空白样品中添加混合标准溶液,从而建立基质匹配标准曲线,线性范围为0.5~20 μg/kg,共设定6个点, 以丰度较强的子离子进行定量分析,丰度次之的子离子进行定性分析,以质谱图峰面积与药物质量分数关系作图,横坐标(X)为药物质量分数(μg/kg),纵坐标(Y)为峰面积,绘出标准曲线。 其中,β-受体激动剂类和喹诺酮类兽药采用内标法确定质量分数,其他种类兽药的质量分数采用外标法进行确定。
1.3.5 准确度和精密度试验在阴性生鲜猪肉制成的肉糜中分别添加1、5 μg/kg 和10 μg/kg 3个水平的混合标准溶液,每个质量分数设6个平行。 以平均回收率(recovery)计算结果评价方法的准确度,根据平行样品的相对标准偏差 (Relative Standard Deviation,RSD)评价该方法的精密度。
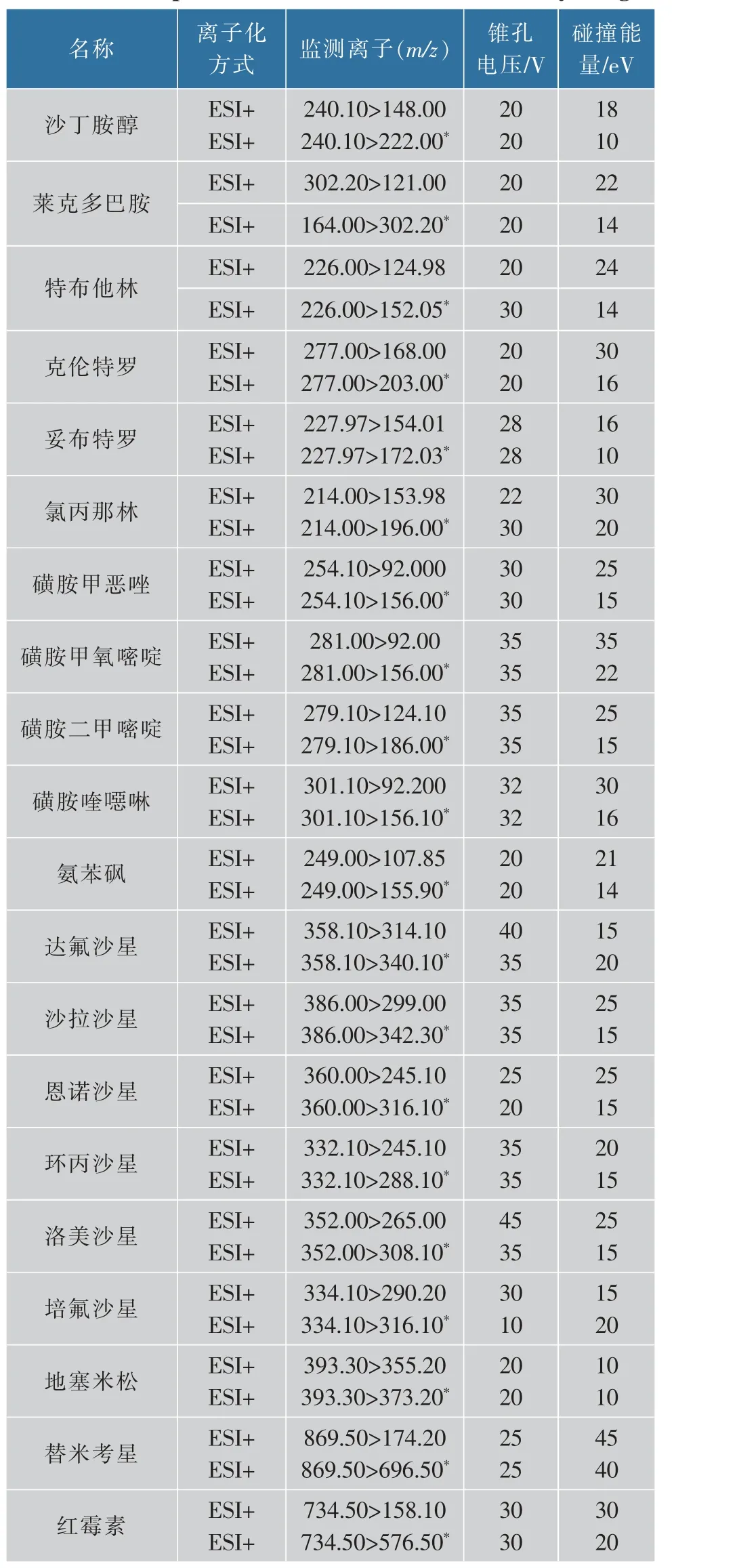
表2 20种兽药质谱条件Table 2 MS parameters of 20 kinds of veterinary drugs
1.3.6 方法的实际应用选用市售生鲜猪肉样品10 份,其中5 份采购自超市,5 份采购自农贸市场,参照1.3.1 和1.3.3 进行样品前处理和上机测定,以验证该方法的实际应用效果。
2 结果与讨论
2.1 提取溶剂的确定
参考相关文献,提取多兽药残留时普遍使用乙腈、甲醇等溶液或者乙腈、甲醇和一定比例其他溶液混合用作提取液。 作者比较了体积分数1%乙酸乙腈、 体积分数0.2%甲酸-80%乙腈水和体积分数80%乙腈水溶液作为提取溶剂时的回收率(加标质量分数为10 μg/kg)。 表3 表明:选用体积分数80%乙腈水溶液作为提取溶剂时,20种药物的回收率均在60%~120%; 以体积分数1%乙酸乙腈溶液作为提取溶剂时,仅有7种药物的回收率在60%~120%;而体积分数0.2%甲酸-80%乙腈水作为提取溶剂时,仅有8种药物的回收率在60%~120%,测定准确性较低。 因此,选用体积分数80%的乙腈水作为提取溶剂。

表3 不同提取溶剂加标回收率的比较Table 3 Comparison of spiked recovery with different extracting solutions
2.2 复溶溶剂的确定
在氮吹完成后进行复溶操作时,复溶溶剂的选择会影响测定的准确度与精密度。 作者在以体积分数80%乙腈水作为提取溶剂的基础上,比较体积分数10%甲醇水、 体积分数15%乙腈水和体积分数0.1%甲酸-50%乙腈水溶液分别用作复溶溶剂时的回收率(加标质量分数为10 μg/kg)。表4 表明:当体积分数10%甲醇溶液作为复溶溶剂时,所有药物的回收率均在60%~120%之间;在体积分数15%乙腈溶液用作复溶溶剂时, 有10种药物的回收率位于该范围内; 而使用体积分数0.1%甲酸-50%乙腈溶液复溶时,仅有9种药物的回收率位于该范围内。因此,选择体积分数10%的甲醇溶液作为复溶溶剂。
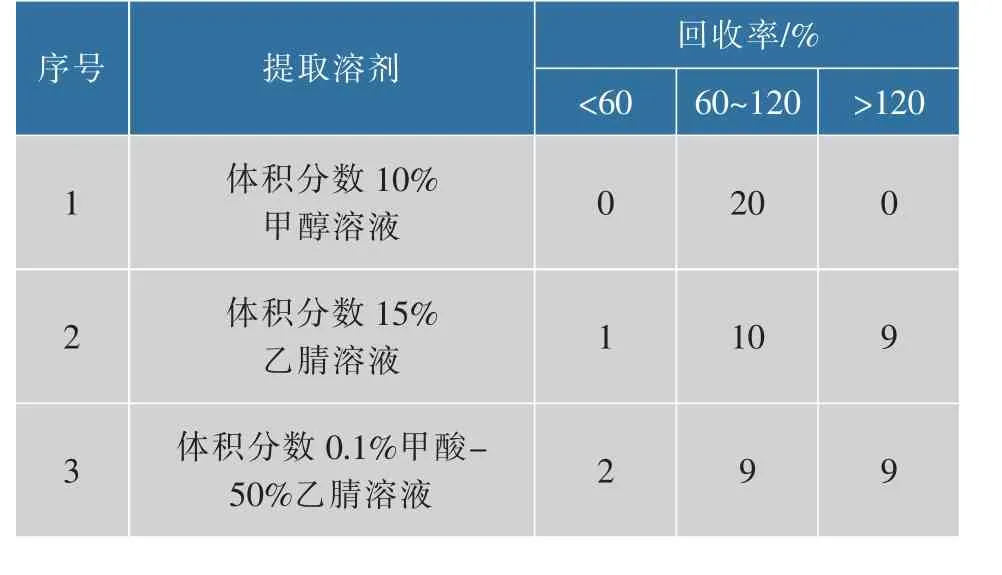
表4 不同复溶溶剂加标回收率的比较Table 4 Comparison of spiked recovery with different dissolving solutions
2.3 标准曲线、相关系数、检出限及定量限
本试验中喹诺酮类和β-受体激动剂类兽药的标准曲线采用内标法绘制,其他种类兽药标准曲线均采用外标法进行绘制,20种药物的线性方程、相关系数、 检出限以及定量限见表5。 20种药物在0.5~20 μg/kg 内呈现出良好的线性关系, 其r 值均大于0.995。 检测的20种药物中,18种药物的检出限为0.5 μg/kg,定量限为1.7 μg/kg;其余2种药物(地塞米松和替米考星) 的检出限分别为1μg/kg 和5 μg/kg,定量限分别是3.3 μg/kg 和16.7 μg/kg。
2.4 方法精密度与准确度
分别在阴性猪肉糜中添加1、5、10 μg/kg 3个质量分数的混合标准溶液,采用UPLC-MS/MS 检测以确定样品中兽药残留的回收率及RSD,且每个质量分数设6个平行。 表6 表明:20种兽药3个不同质量分数的平均回收率在62.1%~118.3%,RSD 位于0.4%~16.7%之间。
2.5 方法实际应用结果
选用10 份市售生鲜猪肉作为供试材料,并设置阳性对照和阴性对照控制样品的质量。 表7 表明:仅有一份超市销售的猪肉样品中检测出磺胺二甲嘧啶,质量分数为9.94 μg/kg。
3 结 语

表5 20种药物线性方程、相关系数、检出限以及定量限Table 5 Linear equations,correlation coefficients,LOD and LOQ of 20 drugs
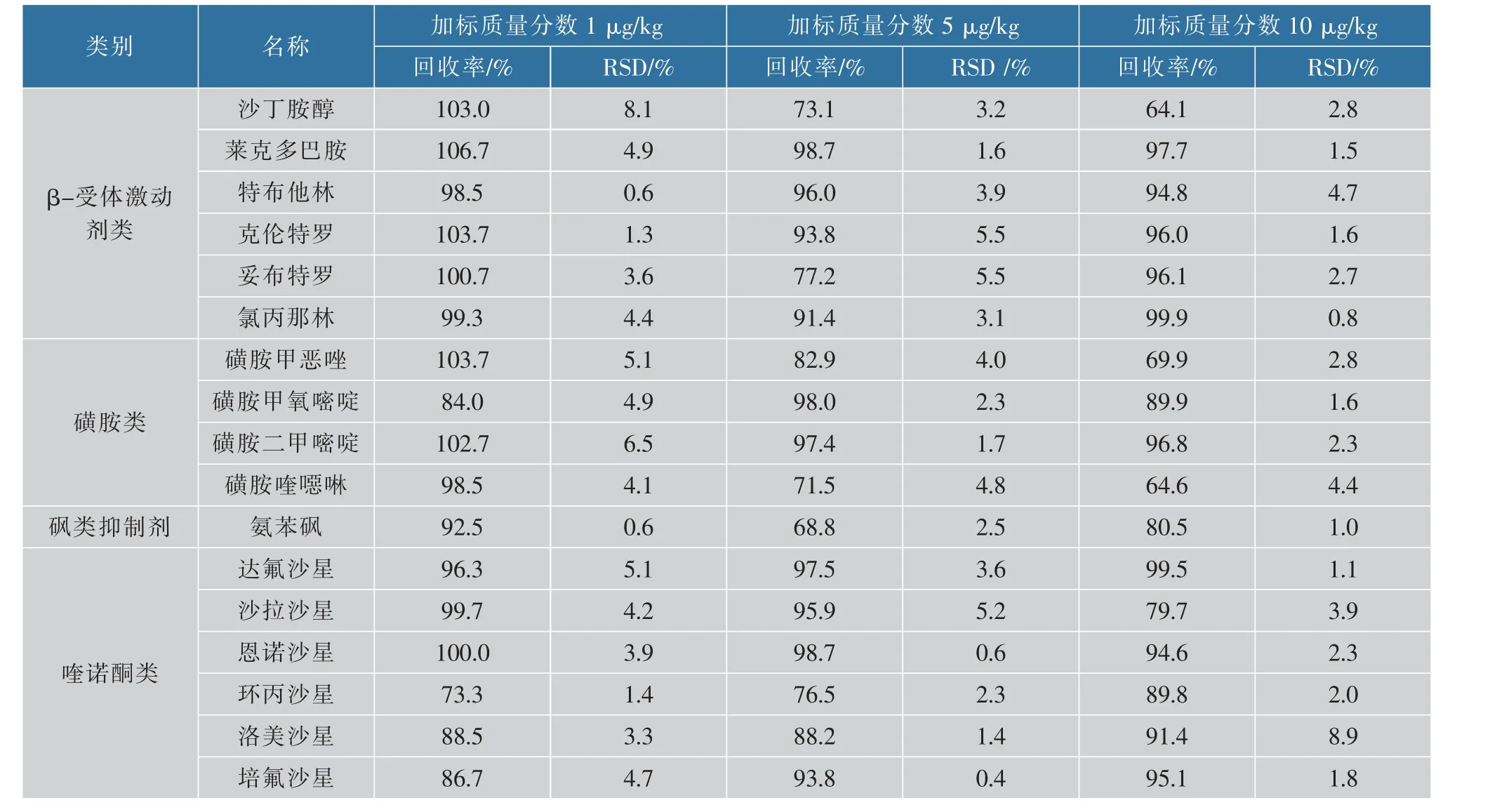
表6 20种药物的加标回收率以及相对标准偏差(n=6)Table 6 Spiked recovery and relative standard deviation(n=6)of 20 drugs
超高效液相色谱-串联质谱法灵敏度好、 准确度高,同时能够突破现行有效检测方法分类检测的局限,实现多种类兽药残留的高通量同时检测。 作者应用UPLC-MS/MS 法检测猪肉中6 类共20种兽药残留,所有检测目标物分离良好,重现性好,并且采用Oasis PRiME HLB 小柱净化样品,使得前处理方法简便快捷,检测效率提高。

续表6
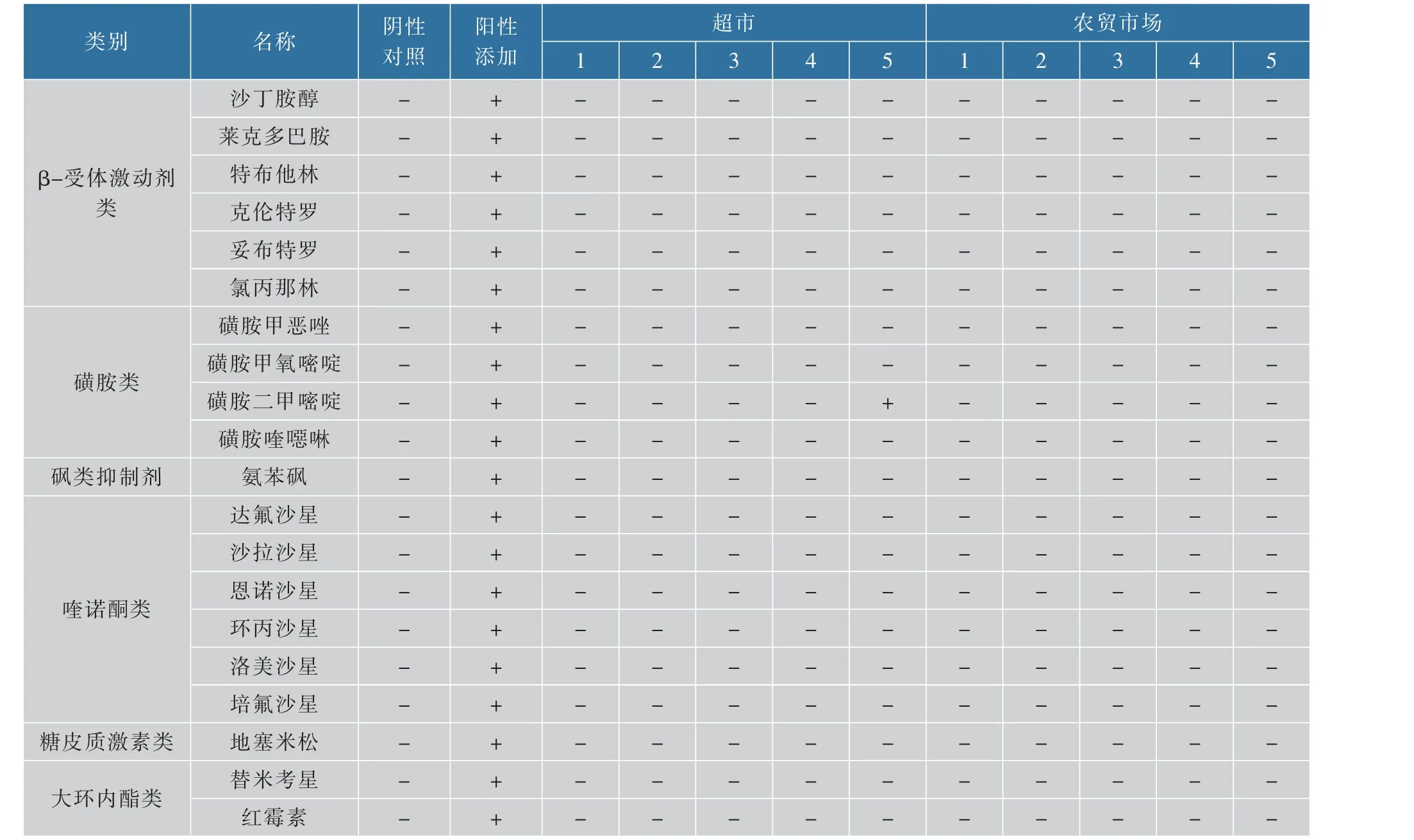
表7 生鲜猪肉20种兽药残留检测结果Table 7 Detection results of 20 drugs in meat
作者试验20种兽药残留的检出限可达到0.5~5.0 μg/kg, 其中喹诺酮类和大环内酯类兽药残留的检出限分别低于国家现行有效标准GB/T 21312-2007 和出入境检疫行业标准SN/T 1777.2-2007 中规定的1~3 μg/kg[25]和20 μg/kg[26];磺胺类兽药残留的检出限能达到现行有效检测方法农业部1025 号公告中规定的0.5 μg/kg[27]。 低、中、高3个浓度水平的加标回收率在62.1%~118.3%, 均在国家现行有效标准GB/T 27404-2008 中规定的60%~120%内[28],RSD 为0.4%~16.7%,表明该方法具有良好的准确度和精密度。 该方法在实际应用中,检测效果良好,可用于猪肉样品中多种兽药残留的高通量快速检测。
