有机无机杂化PVA/SiO2阳离子交换膜的制备及电化学性能表征
2020-09-09李艳红王三反周键李志伟王仲宇李瑞红
李艳红,王三反,周键,李志伟,王仲宇,李瑞红
(1.兰州交通大学 环境与市政工程学院,甘肃 兰州 730070;2.寒旱地区水资源综合利用教育部工程中心,甘肃 兰州 730070;3.牡丹江市自然资源局,黑龙江 牡丹江 157000;4.西安建筑科技大学 建筑设计研究院兰州分院,甘肃 兰州 730030)
采用“双膜三室”电解处理高浓度CoCl2废水时,由于阳极产生的Cl2与阳极液中的H2O反应,生成具有强氧化性的HClO,HClO严重破坏阳离子交换膜,Siu等研究表明[1-2]Nafion具有良好的氧化稳定性,但高昂的价格限制了其应用。Honma等研究表明[3-5],有机无机杂化二氧化硅阳离子交换膜在苛刻条件下能够完成分离任务,有机组分可以增强机械强度和离子传导性,而无机组分SiO2可以提高膜的热稳定性和化学稳定性[6]。因此,本文拟制备选择透过性高的有机无机杂化SiO2离子交换膜。
1 实验部分
1.1 试剂与仪器
聚乙烯醇(PVA)、3-巯丙基三乙氧基硅烷(MPTES)、原硅酸四乙酯(TEOS)、甲醛(37.0%~40.0%)、盐酸(35%~38%)、硫酸(95%~98%)、氯化钾、氯化钠、无水硫酸钠、过氧化氢≥30.0%均为分析纯;实验用水为去离子水。
HJ-6多头磁力加热搅拌器;DHG-9070B智能型电热恒温鼓风干燥箱;FA2204B电子天平;ULTRA plus热场发射扫描电镜;Q150R ES高真空离子溅射仪;VERTEX 70红外-拉曼光谱;PGSTAT128N Autolab电化学工作站;DT-830B数字万用表;ZX21直流多值电阻器;QST EXPRESS深度游标卡尺。
1.2 膜制备
将2 mL的原硅酸四乙酯(TEOS)和10 mL的3-巯丙基三乙氧基硅烷(MPTES)加入到100 mL溶解一定量聚乙烯醇(PVA)的热去离子水中得到均质溶液。用1 mol/L的HCl调节pH为2,在室温下搅拌均匀。将得到的溶胶在60 ℃烘箱中静置脱泡12 h。浇铸到钛板上流延成膜。待膜干燥后从钛板上剥离,浸入甲醛(54.1 g),硫酸钠(150.0 g),硫酸(125.0 g)和水(470.0 g)[7]的混合溶液中交联2 h。将膜用去离子水冲洗干净,浸入H2O2溶液中浸泡8 h,使—SH转化为—SO3H,引入活性基团。制备的膜浸泡在2 mol/L的氯化钠中待测试。
1.3 阳离子交换膜的表征
1.3.1 扫描电子显微镜(SEM) 准备不同比例的PVA/SiO2阳离子交换膜样品,将样品放在液氮中萃断,制备断面测试样品。将准备好的表面和断面样品进行喷金处理后,采用ULTRA plus型扫描电子显微镜观察样品的微观形貌。
1.3.2 傅里叶变换红外光谱(FTIR) 采用VERTEX 70傅里叶变换红外光谱仪测定样品的FTIR图谱,测试的波数区间为4 000~400 cm-1。将制备的样品在60 ℃烘干,磨粉,放入研钵中与溴化钾混合,压片。
1.3.3 吸水率和离子交换容量(IEC)
1.3.3.1 吸水率的测试 剪取面积为2 cm2的膜样若干,浸泡在2 mol/L的NaCl中,25 ℃下搅拌6 h。将膜样取出,用滤纸拭干其表面水分,称重m1。将膜于60 ℃干燥至恒重,取出称重m0。用式(1)计算吸水率η(%):
(1)
式中m1——湿膜质量,g;
m0——干膜质量,g。
1.3.3.2 IEC的测试 用传统滴定技术测量IEC。将制备的PVA/SiO2阳离子交换膜浸入2 mol/L的H2SO4溶液中24 h,转化为H+型。用蒸馏水洗涤除去过量的H2SO4,在2 mol/L的NaCl溶液浸泡24 h,将氢型的膜转化为钠型。用0.05 mol/L的NaOH溶液反滴定来测定IEC值。IEC(mol/g)的计算公式见式(2):
(2)
式中n——用钠离子交换的氢离子的物质的量,mol;
M烘干——膜样烘干后的质量,g。
1.3.4 氧化稳定性 用Fenton试剂(3%的H2O2和3 mg/L的FeSO4)测量制备的PVA/SiO2膜和商品苯乙烯阳离子交换膜的氧化稳定性。将4种膜分别裁剪成为10 cm×10 cm的矩形方块(共6块,其中3块用于80 ℃下测试,另外3块用于40 ℃下测试),在60 ℃烘干,记录重量。将这些膜浸泡在装有一定量的Fenton试剂中,在80 ℃下和40 ℃(实际温度)下进行氧化。一定时间后记录浸入前和浸入后的干膜的重量损失。通过式(3)计算重量损失:
(3)
其中,W1是氧化前的干膜重量,W2是氧化后的干膜重量。
1.3.5 膜电阻、迁移数和选择透过性
1.3.5.1 膜电阻 在恒温25 ℃的条件下,将合成的膜置于装有2 mol/L NaCl溶液的电解槽之间,采用电化学工作站完成测量工作。制备的PVA/SiO2阳离子交换膜面积电阻,采用文献[8]描述的方法计算电位差。面积电阻计算如下:
Rm=R1-R2
(4)
其中,Rm为单位膜面积电阻;R1为含有离子交换膜时的电阻;R2为没有离子交换膜时的电阻。
r=(RmA)
(5)
其中,r为阳离子交换膜的面积电阻;A为阳离子交换膜的面积。
1.3.5.2 迁移数[9]迁移数表征了离子交换膜对反离子的选择透过能力,即对同离子的排斥能力,通常用通过离子交换膜所移动的离子的当量百分数来表示。一般通过测定静态下的膜电位而计算,膜电位的测定如下:将用1.5 mol/L的KCl溶液预处理的待测膜固定在文中[9]所示的测试槽中,在膜的两侧注入不等浓度的氯化钾(KCl)溶液(C1=1 mol/L,C2=2 mol/L,C1/C2=2∶1,25 ℃)。在实验期间,通过连接两隔室,并使用数字万用表(DT-830-B)和甘汞电极(通过KCl盐桥)测量跨膜电位。重复测量,直至获得恒定的电位。通过膜电位计算迁移数。
(6)
式中E测——特定温度下测量得到的膜电位,mV;
E0——特定温度下的理论电位值,mV;
t+——测得的阳离子交换膜的迁移数。
1.3.5.3 选择透过性 离子交换膜的选择透过性用迁移数来表示,见式(7):
(7)
其中,t是溶液的迁移数。在25 ℃,NaCl溶液的迁移数是39%。
2 结果与讨论
2.1 膜制备
在pH为2的酸性条件下,MPTES和TEOS发生水解和部分缩聚后生成硅醇。硅醇发生脱水缩聚反应产生交联键,交联键不仅与聚合物网络键合,而且还与二氧化硅部分键合。二氧化硅与聚合物网络之间的共价键增强了有机相和无机相之间的界面相互作用。在该过程中,MPTES和TEOS的水解缩聚形成了具有三维网络的Si—O—Si键,从而提高了有机无机杂化PVA/SiO2膜的氧化稳定性,同时,Si—O—Si键的产生使制备的膜具有良好的成膜性能和机械性能[10]。PVA/SiO2膜的形成机理见图1。制备的溶胶和离子交换膜见图2。

图1 有机无机杂化PVA/SiO2膜制备机理

图2 制备的溶胶凝胶和制备的PVA/SiO2膜
2.2 微观形貌
4%PVA,6%PVA和8%PVA的PVA/SiO2表面和断面形貌,见图3。
由图3中(a)、(b)和(c)可知,随着PVA质量分数的增加,杂化膜表面的裂纹和褶皱逐渐消失。在相同的放大倍率下,4%PVA杂化膜表面的褶皱和裂缝密度大,裂缝明显且深,表面的颗粒少;6%PVA杂化膜表面的褶皱减少,无裂缝,裂纹可见,表面颗粒增多;8%PVA杂化膜表面无褶皱和裂缝,有均匀的纹路和团聚。这主要是因为PVA自身脱水缩聚。随着PVA质量分数的增加,硅烷的质量分数相对减少,硅烷交联反应更不易发生,更多的是PVA自身脱水缩聚反应(宏观上表现为褶皱减少,PVA缩聚产生团聚部分增多),这也能够解释(b)中的褶皱减少而颗粒增加,而(c)中的褶皱完全消失,能够看见团聚的颗粒和突起。此外,3张杂化膜断面(a’)、(b’)和(c’)均匀密实,没有任何的裂纹与损坏,可能是由于杂化膜的内部没有相分离。4%PVA杂化膜的表面(a)有裂缝,截面(a’)没有裂缝和损坏,可能是由于在进行SEM电镜测试前,将杂化膜在60 ℃烘箱中干燥,水分的蒸发造成了严重的表面龟裂。

图3 PVA/SiO2和苯乙烯膜的表面和断面形貌
由图3a~3d可知,在相同的放大倍率下,苯乙烯膜表面有很多小孔,而制备的PVA/SiO2膜表面致密。同时,4张膜(a’,b’,c’和d’)的横截面也可以看出苯乙烯膜具有明显的孔隙和空隙,能够证明商品苯乙烯膜的选择透过性比制备的PVA/SiO2膜低的主要原因是由于大孔隙的存在导致了离子的泄露。因此,这也可能是制备的PVA/SiO2膜的选择透过性高于苯乙烯膜的主要原因。
2.3 FTIR研究
记录3种制备的杂化膜的FTIR图谱见图4。
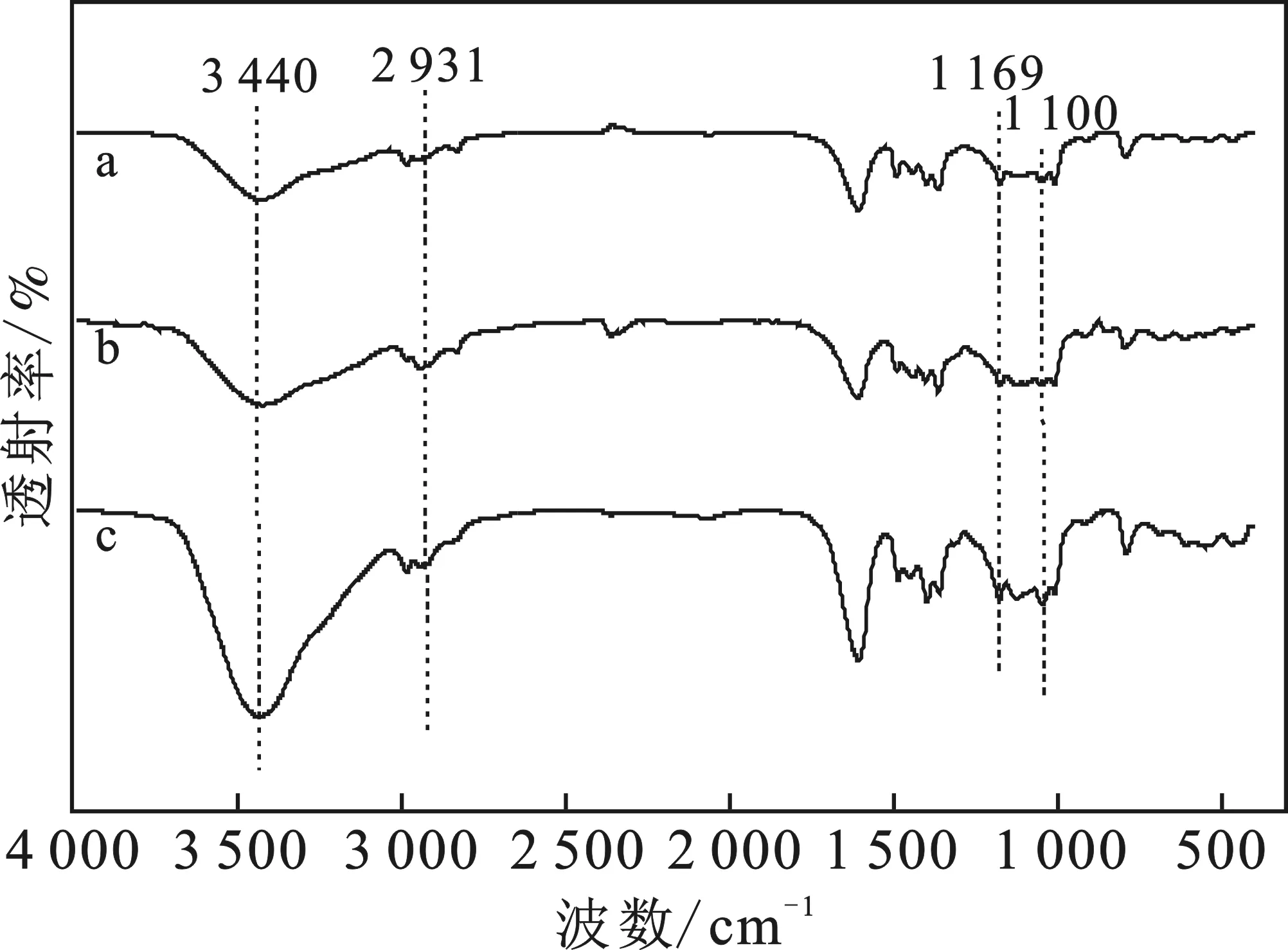
图4 有机无机杂化PVA/SiO2膜的FTIR图谱
由图4可知,O—H的伸缩振动峰在3 200~3 650 cm-1[11]产生吸收峰,谱带较强、较宽,它可以作为判断有/无机醇类、酚类和有机酸类的重要依据。因此,(a)3 440 cm-1、(b)3 435 cm-1和(c)3 435 cm-1有可能是交联过程中产生的C2H5OH和H2O 对应的O—H伸缩振动峰。饱和C—H伸缩振动峰出现在3 000 cm-1[11]以下,约在2 800~3 000 cm-1范围内产生吸收峰,因此,(a)2 984,2 928 cm-1、(b)2 937,2 828 cm-1和(c)2 990,2 926 cm-1可能是饱和C—H伸缩振动峰。还可以看出,大约1 200 cm-1[7,10,12-14]吸收带都很强,这些图谱可能包括在交联过程中形成的Si—O—Si,Si—O—C,又由于—SO3H基团的特征峰位于约1 100 cm-1和1 200 cm-1区域,因此,Si—O—Si,Si—O—C和SO3H的伸缩带重叠,在图中无法区分。在2 600~2 550 cm-1处没有出现S—H对应的峰,说明巯基全部转化为—SO3H。同时,IEC的存在也能够证明交联过程中SO3H基团的形成。
2.4 氧化稳定性
以6%PVA/SiO2和苯乙烯膜为测试对象。在80 ℃下氧化3 h后发现制备的PVA/SiO2膜完全溶解,苯乙烯膜的重量损失为61.17%。 40 ℃下的氧化实验显示,6%PVA/SiO2膜和苯乙烯膜氧化12 h的重量损失都很低,且膜的结构都完好无损,说明高温加速了PVA/SiO2膜的氧化,同时也说明实际中“双膜三室”高浓度CoCl2电解温度(40 ℃)下,PVA/SiO2膜可以替代商品苯乙烯膜。对于基于非氟烃骨架的6%PVA/SiO2聚合物电解质材料,这种氧化稳定性实际上已经非常高了。主要原因在于:(1)交联增加了聚合链的堆积密度,增加了对自由基攻击膜的阻抗性;(2)交联对聚合物材料机械强度的增强作用,交联等价于聚合物分子量的增加。当聚合物链被自由基攻击时,破碎的分子链仍然可以通过交联点附着在聚合物网络上,这延长了在自由基攻击下聚合物骨架完全破碎的时间。然而,与苯乙烯膜相比,6%PVA/SiO2膜在80 ℃下的氧化稳定性仍然相对较差。因此,需要通过大量的工作提高其氧化稳定性。
2.5 阳离子交换膜的电化学性质
2.5.1 吸水率和离子交换容量 离子交换膜的吸水率受其化学结构和与其接触的电解质溶液的性质和浓度的影响[13]。3种制备的PVA/SiO2膜和苯乙烯膜的吸水率见图5。

图5 离子交换膜的IEC、吸水率、膜电阻和膜厚度
由图5可知,随着PVA质量分数的增加,PVA/SiO2膜的吸水率也随之增加。一般而言,具有一定程度组成的膜吸收相同量的水,其中可离子化基团的密度在膜基质中是相同的[15-16]。亲水性PVA含量的增加是离子交换膜吸水率增加的主要原因。
离子交换容量提供了膜中存在的可离子交换基团的指示[17]。膜的离子交换容量(IEC)对质子的传导性和吸水率起着重要的作用,这取决于亲水性官能团的密度[18],也是离子交换膜中产生离子电导率的原因[13]。由图5还可知,制备的膜IEC在0.61~1.17 mol/g范围内。IEC随着PVA含量的增加而增加,主要原因在于PVA含量的增加在一定程度上改善了膜基质结构。因此,可以推断,PVA含量的增加,离子交换膜的物理化学性质得到了改善,因此磺化度增强[10,12-13,19]。
2.5.2 膜电阻、迁移数和选择透过性 在测试制备的PVA/SiO2和苯乙烯膜电阻之前,将待测的膜在2 mol/L的NaCl溶液中浸泡24 h以上。膜电阻的测试数据见图5。了解膜面积电阻对于评估不同官能团的作用非常重要[20]。实验研究表明,阳离子与固定电荷之间的相互作用,移动离子的相对大小以及膜结构特性是影响离子交换膜电阻的3个关键因素[21-22]。由图5可知,6%PVA/SiO2膜的面积电阻最低,可能原因是由于膜的厚度较低(0.11 mm)。PVA/SiO2膜的面积电阻高于Nafion膜[23],但低于苯乙烯膜,因此,在电解使用过程中能耗比苯乙烯膜低,利用PAV/SiO2膜电解处理高浓度的CoCl2废水比苯乙烯更经济。
用离子交换膜分离不等浓度的电解质时,由于共离子和反离子的迁移率不同,在膜上产生膜电势。膜电位的大小取决于离子交换膜的电特性以及电解质的特性和浓度[24]。
由图6可知,制备的PVA/SiO2膜的选择透过性和迁移数都高于苯乙烯膜,且随着PVA质量分数的增加,制备的膜选择透过性和迁移数都减小,即选择透过性和迁移数从高到低的顺序是:4%PVA/SiO2>6%PVA/SiO2>8%PVA/SiO2。迁移数和选择透过性的规律一致。迁移数和选择透过性的变化可能受到固定电荷密度和孔密度的变化[13]。固定电荷密度的变化增强了共离子的Donnan排斥作用。因此,8%PVA/SiO2的选择透过性较低,可能因为孔密度变化较大。IEC和吸水率测试的结构证实了这一假设。
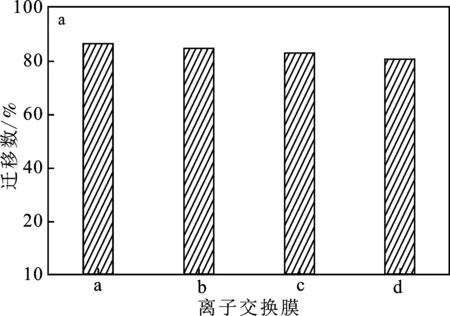
图6 有机无机杂化PVA/SiO2膜和苯乙烯膜的迁移数和选择透过性
3 结论
(1)采用溶胶凝胶法制备了有机-无机杂化PVA/SiO2膜,并通过氧化—SH基团引入—SO3H基团。TEOS和MPTES直接添加到PVA浇铸溶液中,在酸性条件下(pH=2)形成膜时,浇铸液中的TEOS和MPTES可以水解和缩聚,形成Si—O—Si聚合物,然后制备有机-无机杂化膜。
(2)制备的PVA/SiO2膜具有比苯乙烯膜高的选择透过性和低电阻。尤其是6%PVA/SiO2可能适用于“双膜三室”钴回收工艺,作为苯乙烯膜的替代膜。但制备的PVA/SiO2膜在高温下的氧化稳定性能较差,仍然需要进一步的研究。
