电化学氢-水转化系统中电解水和氢燃料电池催化剂的设计
2020-09-03彭立山魏子栋
彭立山,魏子栋
Chongqing Key Laboratory of Chemical Process for Clean Energy and Resource Utilization, School of Chemistry and Chemical Engineering, Chongqing University, Chongqing 400044, China
1. 引言
目前全球能源消耗持续增长,但近88%的能源经济依赖化石燃料[1]。尽管化石燃料在能源组合中所占份额过大,但化石燃料时代或许即将结束[2]。化石燃料彻底耗尽或其开采变得无利可图只是时间问题。除了供应量减少的问题外,化石燃料的使用对全球生态系统构成了重大风险。目前,世界上的能源供给主要依靠化石燃料的燃烧。这种燃烧所产生的副产品(如二氧化碳、氮氧化物、硫氧化物和细颗粒物等)严重污染空气、土壤和水[3–5]。我们迫切需要采用新的思维方式,以便找到解决这些问题的方案,并设计更安全、可持续的能源供应系统。可再生能源将在世界能源的未来中发挥至关重要的作用。然而,可再生能源和传统能源之间的巨大差异造成了市场壁垒[6–9]。可再生能源产生的能量(主要是电力形式)可能在短时间内发生不可预测的变化。例如,太阳能系统仅在阳光照射时产生能量。其他可再生能源,如风能和潮汐能,同样具有不稳定性这一不利因素[10]。这种不稳定性使当前的可再生能源发电的可靠性低于化石燃料产生的能源,因为其输出强度依赖于天气条件(即云或风)和时间(即白天或晚上)。为了使可再生能源大规模应用,需要高效的电能转换和高密度的电能存储技术,以实现能源分配。
电化学氢-水转化(H2+O2⇌ H2O)是一种清洁高效的可持续能源系统执行解决方案[11,12]。具体来说,可再生能源可以通过水电解转化为储存在氢气中的化学能[13]。相反,氢分子可以通过电化学的方式重组成水,以便通过燃料电池输出电能。在该能量系统中,氢充当能量载体,并且能量转换与热循环无关[1,14,15]。因为该系统是基于氢和水的电化学反应,可以有效地避免对自然环境和人类健康有害的气体和化合物的释放。在实际应用中,氢气必须首先被获取,然后被储存,最后被转换回水以释放储存的能量[16]。为了实现这一目标,高效、低成本的水电解和燃料电池技术必须有效地结合起来。电化学过程是这些能量转换技术的核心,包括水电解技术中的析氢反应(HER)和析氧反应(OER),氢氧燃料电池中的氧还原反应(ORR)和氢氧化反应(HOR)[17,18]。这四种电化学反应的效率对上述能量转换技术的输出性能有很大的影响。因此,在该可持续能源系统中,最关键的问题是如何在催化电极表面有效地催化以上反应,以获得最低的过电位和最高的电流密度[19–22]。除了电化学反应引起的电压降外,内阻和传质电阻等也会影响水电解和燃料电池的总电压。因此,通过优化电极结构来加速电子、质子的转移和产物的脱附是另一个需要关注的问题。
本文对用于电化学氢-水转化电催化剂的结构工程的最新进展进行了全面综述。本文主要讨论了两个问题:①电化学氢-水转换系统能量耗散的来源;②基础科学与实用技术相结合驱动高能量转换效率电催化剂的结构设计。第2节中,在简要介绍了氢-水转化过程中的电化学过程之后,我们从实用的角度回顾了水电解和燃料电池两种功能技术的能量耗散,并利用经典动力学分析了催化剂表面发生电化学反应的关键障碍。借助于反应中间体之间的标度关系,我们构建了一个了解催化性能趋势的框架,为开发用于广泛反应的高效催化剂提供指导。第3节总结了设计高性能电催化剂的通用策略,并讨论了它们的优缺点。这部分介绍了通过结构设计实现的高效电催化剂的典型案例,展示了合成化学、电催化化学和计算化学的有机结合。最后概述了电化学氢-水转化系统中的关键科学问题,为高效、可再生能源系统的催化剂设计提供了方向。
2. 电催化基础
2.1. 氢-水转化中的电化学反应
如图1所示,该可再生清洁能源系统涉及两种不同的功能技术:电解水和燃料电池。这两种技术的电解槽主要由四部分组成:电解液(如H2O)、离子交换膜(如Nafion膜)、阳极电极和阴极电极[23,24]。为了提升这两种技术的效率,阴阳电极表面一般涂覆着高效稳定的催化剂层。在水电解槽中,电能被消耗以将水分解为气态氢(H2)和氧(O2)。以酸性水电解为例,当电子通过外回路时,质子通过离子交换膜进入阴极,与电子结合形成氢分子,水在阳极氧化形成氧分子和质子。
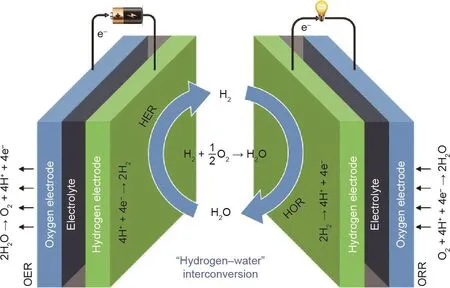
图1. 氢-水的电化学转换中电解水和燃料电池的反应原理图。
燃料电池中发生的电化学反应与水电解过程完全相反[25]。氢和氧的自然“冷”燃烧发生在燃料电池装置中,其中氢作为燃料,氧作为氧化剂。通常,氢通过电极孔扩散到阳极表面。通过催化剂层的催化作用,吸附的氢被电离并在电极上释放出一个电子。接下来,通过电解液的氢离子和通过外部电路的电子都到达阴极,与氧分子重新结合形成水分子,并释放出电流。通过使用合适的水或空气冷却系统,可以消除内部反应和电阻产生的热量。表1总结了水电解和燃料电池在不同介质中的半电池反应。这四个反应可分为两个可逆反应对:相对于可逆氢电极(RHE),平衡电位(U0)为0 V的HER和HOR,以及U0=1.23 V的ORR和OER。从化学角度来看,氢-水转化是由两个氧化还原对组成的:高电位的水/氧对和相对低电位的水/氢对[26]。在酸性介质中,水合质子将电荷从阳极转移到阴极,而在碱性电解质中,氢氧化物离子作为电荷载体从阴极转移到阳极。
2.2. 水电解和燃料电池的能量耗散
根据氢-水转化的反应热力学,水电解和氢氧燃料电池在标准条件下具有相同的起始电位(1.23 V)。然而,这两种技术的实际起始电位与标准电位相差甚远。在实际操作条件下,即使使用最先进的贵金属作为电催化剂,燃料电池的电压始终低于0.9 V,而水电解的运行电压往往高于1.8 V [2,27–29]。在实际应用中,为了驱动电化学反应过程,必须克服许多势垒,包括电路的电阻、电化学反应的活化能、产物气泡或水对电极表面的阻塞,以及电解质溶液的离子转移电阻等[24]。这些势垒需要足够的电能供应以克服,这大大降低了能量转换效率,导致工作电位低于热力学电位,即发生所谓的极化(或过电位,或过电压)现象[30]。图2示出了典型的液体电解质电池中的电阻(即势垒)[31,32]。两端的第一个电阻(RaΩ和RcΩ)是外部电路电阻,包括阳极和阴极的导线和连接的电阻以及电子穿过催化剂层的电阻(这些材料通常不是良好的电子导体)[33]。Ract和Rcct起源于阴阳极表面半电池反应的过电位[34]。RaD和RcD是靠近电极表面的扩散层引起的传质电阻[35]。在水电解中,由于产生的气泡对电极表面的覆盖,阻碍了电极与电解液的接触。类似地,产物水在电极与反应物氧气之间起到阻塞作用,导致碱性燃料电池(AFC)中的传质阻力。Rions源于电解质中离子的传输阻力,Rsep源于电池隔膜的阻力[36]。类似的电阻情况同样出现在其他类型的电池中,如零极距电解池和质子交换膜(PEM)电池[37,38]。
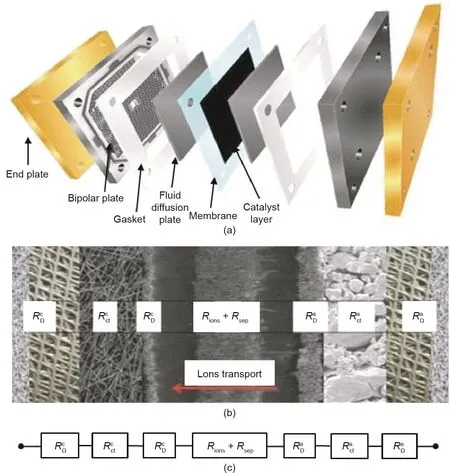
图2. (a)典型的液态碱性燃料电池示意图;(b)电解池的横截面;(c)等效电路[31]。
以上电池系统中的电阻可分为三类:活化电阻(电化学反应引起的损耗)、欧姆电阻(离子和电子传导引起的损耗)和浓度电阻(物质传输引起的损耗)。这三类电阻共同决定了电化学电池的电流-电压(i-V)曲线的特征形状[39,40]。图3显示了水电解和燃料电池的典型i-V曲线。在水电解的i-V曲线中[图3(a)],当电压高于热力学电解电压1.23 V后,电流开始流过电解池。在低电流密度下,欧姆电阻引起的电压降很小,反应活化过电压占电压降的主要部分。此时极化曲线的对数形状(Tafel区域)归因于阳极和阴极的电荷转移现象。随着过电压的进一步增大,反应活化势垒减小,极化曲线呈线性。这种线性形状表明此时欧姆电阻是电解池的最关键动力学参数。在燃料电池的i-V曲线中[图3(b)],活化电阻主要影响曲线的初始部分,欧姆电阻的影响主要在曲线的中间部分,浓度电阻的影响在曲线的尾部显著。尽管这两种功能技术中发生的反应是可逆的,但i-V曲线的形状并不相同:水电解的i-V曲线在高电位下通常遵循Butler-Volmer模型;而由于对传质速率的限制,燃料电池的i-V曲线往往在高电位下显示恒定值。
为了提高两种技术的能量转换效率以改善能源系统的整体性能,必须了解上述电阻产生的缘由,以便将其最小化。欧姆损耗是由电极材料对电子流的电阻和电解液对离子流的电阻引起的。这些损耗可以通过使用高导电材料作为布线和电极基板,以及通过减小两个电极之间的距离来降低[41–43]。通过增加气态反应物的压力或液体电解质的浓度,可以减轻由传质引起的损耗[44,45]。这两种电压降主要取决于电池的设计和运行条件。除了上述两类电阻外,电化学电池中的大部分电压降(>60%)是由半电池反应的吉布斯自由能变化引起的[39]。根据反应的方向,活化极化大大增加发生氧化反应的阳极电压,并降低了发生还原反应的阴极电压。
在电化学中,Butler-Volmer关系被用作主要出发点,将催化剂-电解质界面上的过电压(η)与该界面上的电流密度j(A·cm–2)联系起来[46]:
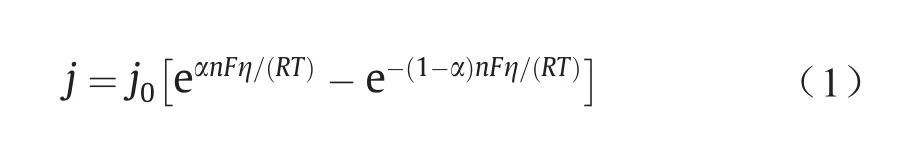
式中,η是过电压,即界面上的实际电压与平衡电压之差;j0是交流电流密度,单位为A·cm–2;α是电荷转移的系数;n是在电化学反应中转移的电子数;F≈96 485 C·mol–1,是法拉第常量;R是摩尔气体常量(0.082 J·K–1·mol–1);T是热力学温度(K)。Butler-Volmer方程表明,电化学反应产生的电流随过电压和交换电流密度呈指数增加。实际上,j0表示反应物与产物处于平衡状态时的“交换速率”,是提高反应速率的重点。为简单起见并考虑浓度效应,正向反应下j0的定义为:
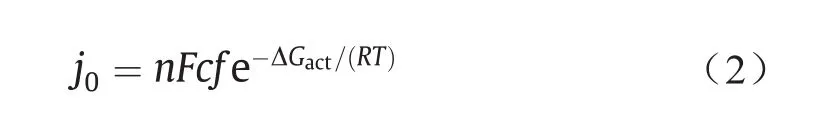
式中,c是反应物的表面浓度;f是产物的衰减率;ΔGact是正向反应的活化势垒。等式(2)清楚地表明,在给定的环境条件下,减小活化能垒(ΔGact)将增加j0。在实际反应中,只有处于活化状态的物质才能经历从反应物到产物的转变。催化电极是物质活化和转变的场所,反应的活化能极大程度上取决于电极材料[47]。因此使用高性能的催化电极可显著降低反应的活化势垒,进而显著提高j0。基于对活化能、电极材料和表面构型之间关系的理解,现今大量的研究工作集中于高效催化电极材料的设计以降低电极反应的活化能。
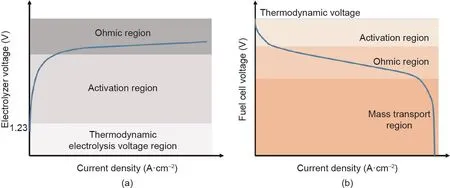
图3. 水电解(a)和燃料电池(b)的极化曲线图。
2.3. 用于电化学氢-水转化的催化剂设计指南
根据反应机制,反应活化势垒(ΔGact)可以用平衡电势下速率决定步骤(RDS)的自由能变化(ΔGmax)来定量,并且其在不同催化材料上的理论值可以通过密度泛函理论(DFT)来计算。通过绘制j0与ΔGmax之间的火山曲线,可以建立活化能与电极材料之间的关系。最常见的火山曲线是基于Langmuir吸附类型的析氢反应速率描述,其最大值位于氢吸附自由能(ΔGH*)为零附近[48]。在析氢反应中,反应物首先吸附在催化剂表面形成反应中间体(M-Hads)。在上述Volmer步骤之后,氢分子可以由电解液中电子和质子通过Heyrovsky步骤耦合形成,或者通过Tafel步骤直接结合形成[30,49,50]。因此,氢吸附自由能(ΔGH*)是析氢反应速率的决定性因素[51,52]。近年来,DFT计算得到的材料表面的氢吸附自由能被广泛用作许多传统金属和非金属催化材料的活性描述符[53–55]。如图4(a)所示,不同金属的析氢交换电流密度存在显著差异,位于火山曲线顶部附近的金属(如Pt)具有最佳的ΔGH*[56]。如果催化材料对氢的吸附力较弱,氢原子在材料表面几乎不能被吸附,整个反应速率由氢的吸附步骤(Volmer步骤)决定。反之,氢原子在催化材料上的吸附太强,M–Hads键则很难被打断而形成H2,反应决速步为解吸步骤(Heyrovsky/Tafel)。作为HER的逆过程,HOR的RDS是H2在催化剂表面的解离吸附,它涉及电子从催化剂表面转移到H2分子的反键轨道(σ*轨道)[57]。因此,M–Hads的强弱在HOR的动力学中也起主导作用,并且由于这两个反应的高度可逆性,HOR在贵金属表面上的活性趋势与HER相同[图4(b)][58–61]。
与氢参与反应相似,j0和ΔGmax之间的关系也应用于氢-水转化过程中氧参与的反应。如图4(c)、(d)所示,除了决定反应速率的反应中间体不同,氧参与反应的火山曲线形状非常相似。ORR包括以四电子途径将氧还原为水,或以两电子途径生产过氧化氢[62]。事实上,一个直接的四电子ORR反应机制可以是一个解离或结合过程,这取决于催化剂表面的氧解离能垒[63]。因此,ORR火山曲线以氧吸附自由能(ΔGO*)与催化活性相关联[图4(c)][63,64]。对于与氧结合太强的金属,ORR反应速率受到O*或OH*物种脱附的限制。对于与氧结合太弱的金属,反应速度可能受O2中O–O键分裂的限制(解离机制),或者受电子和质子转移到吸附的O2的限制(结合机制),具体情况取决于外加电位[63]。如图4(c)所示,即使铂(Pt)也不在绝对峰值,ΔGO*略低于铂的金属应具有更高的氧还原活性。根据上述热力学火山曲线,Nørskov等[65,66]考虑到氢氧结合能是变化的,建立了ORR的微观动力学模型。他们发现了一种与热力学活性火山曲线非常一致的动力学火山曲线,并确定了ΔGO*比Pt(111)弱0.1 eV的催化剂具有最佳的四电子ORR活性。
OER火山曲线始于1984年,具有很长的历史。当时Trasatti用金属氧化物中金属从低氧化态到较高氧化态的转变焓来描述氧化物电极的OER电催化活性[67]。这项开创性的工作将OER过程视为表面配位化合物的两种不同构型之间的过渡。因此,所有难氧化或易氧化的金属氧化物对OER催化都不是很活泼。难氧化意味着中间体的吸附性较弱,水离解是RDS。反之,易氧化性表明中间产物具有很强的吸附性,则O*或OH*的脱附为RDS。因此,与ORR相同,OER反应速率与ΔGO*有关[68,69]。然而,四电子OER涉及多个中间体(OOH*、OH*和O*),它们的结合能紧密相关且几乎不解耦[63,70],用ΔGO*作为OER活性的单一描述符并不准确。不同的反应中间体的结合能之间存在线性标度关系,即如果与一个反应步骤相关的能量发生变化,其他反应步骤的能量也发生变化。Man等[70]将两个中间产物(ΔGO*–ΔGOH*)的结合能之差作为化合物(包括金红石、钙钛矿、尖晶石、岩盐和方铁锰矿氧化物)催化活性的描述符[图4(d)],其活性很好地服从火山曲线。在金属氧化物材料中,各吸附位点的OH*和OOH*结合能(无论是在OER还是ORR中)均以大约3.2 eV的恒定能量值相互关联[70,71]。由于OOH*和OH*之间的非理想标度,即使对于OER和ORR火山曲线顶部的催化材料,催化剂也存在最小理论超电势(0.3~0.4 V)[63,72,73],包括最优的RuO2析氧催化剂[70]和Pt基ORR催化剂[74]。
火山曲线恰当地证明了Sabatier理论[75],即理想的催化剂与反应中间体结合既不太弱也不太强。换言之,对反应中间体具有适当结合能的催化剂表面可实现最佳催化活性。具体来说,最理想HER/HOR催化剂是具有最小ΔGH*绝对值的材料,理想ORR和OER催化剂则具有优异的ΔGO*和ΔGO*–ΔGOH*。除了降低活化势垒外,还有另一个显著的方法来增加j0,即增加单位面积内反应位点的数量[76–79]。j0表示单位面积的反应电流,而电流密度的面积通常基于电极的几何面积。表面极其粗糙的电极的真实电极表面积可以比其几何电极面积大几个数量级,因此可以提供更多的反应位点。因此,粗糙电极表面的有效j0将远大于光滑电极表面的有效j0。提高活性位点密度的另一个简单方法是增大给定电极上催化剂的用量。然而,过量的催化剂会阻碍电极表面的电荷和质子转移。因此,电极的活性不随催化剂用量的增加而线性增加。
总之,提高电催化剂体系的活性(或反应速率)一般有两种策略:①提高每个活性位点的本征活性;②提高给定电极上活性位点的数量。两种方法各有利弊。不同催化剂的本征活性差异可能超过10个数量级,而由催化剂负载引起的活性差异仅为1~3个数量级。提高每个活性位点的本征活性是实现高活性的最根本、最有效的途径,但其实现必须建立在对反应机制和材料性能深入了解的基础上。增加活性位点的数量是一个更简单的策略,但活性增长是有限的。同时,通过提高催化剂负载量来提高活性需要以增加电极成本和传质阻力为代价。在实际应用中,以上两种策略可以同时实施,从而大大提高催化剂的活性。
3. 电化学氢–水转化中催化材料设计
3.1. 纳米构筑
众所周知,催化剂的电流密度随着活性中心密度的增加而增加。暴露更多的活性中心对获得高催化活性十分重要。纳米构筑被认为是提高活性位点密度最直接有效的策略[80–84]。在过渡金属合金体系中,人们首先认识到实际活性表面积与电催化剂整体性能之间的关系。早在1982年,Brown等[85]发现合金表面通常比单一金属表面粗糙,可以为催化反应提供更多的活性中心。借助于Ni-Mo合金纳米结构化和钼的选择性腐蚀[85–87],Ni-Mo合金的表面积大大增加,催化活性明显提高。近十年来,随着合成技术的快速发展,一系列不同形貌的电催化纳米材料相继问世,包括纳米笼、纳米纤维、纳米花、纳米泡沫、纳米网、纳米针、纳米环、纳米壳、纳米线等[77,88–91]。Faber等[92]报道了金属二硫化钴(CoS2)作为一种高活性催化剂,并证明了几何结构在决定其整体催化性能中的关键作用。与常见的纳米颗粒和纳米薄膜形貌的电极相比,微纳米结构电极的高活性比表面积显著改善了其催化性能(图5)。因此CoS2纳米线电极只需低至145 mV的过电位以驱动–10 mA·cm–2的析氢电流密度。此外,通过促进物质传递和产物(气泡或水)从催化剂表面的脱除,纳米结构具有提高操作稳定性和反应速率的双重功能。Peng等[93]使用自组装和预成形策略,可控合成了具有二维(2D)层状结构的Mo2C/C催化材料。高分散的Mo2C纳米颗粒及二维层状结构有效地促进了Mo2C活性中心的质子和电荷转移,促进了电化学HER过程。此外,我们还进一步合成了一系列三维纳米催化材料,包括NiCo2(SOH)x纳米花[94]、珊瑚状FeNi(OH)x[95]、Ni-VC纳米丛[96]、Ni-Mo2C纳米线[97]和Ni(OH)2@Ni2P纳米柱[98]。所有这些材料都具有高活性表面、快速电子转移和气体逸出通道,有利于催化水电解反应进行。
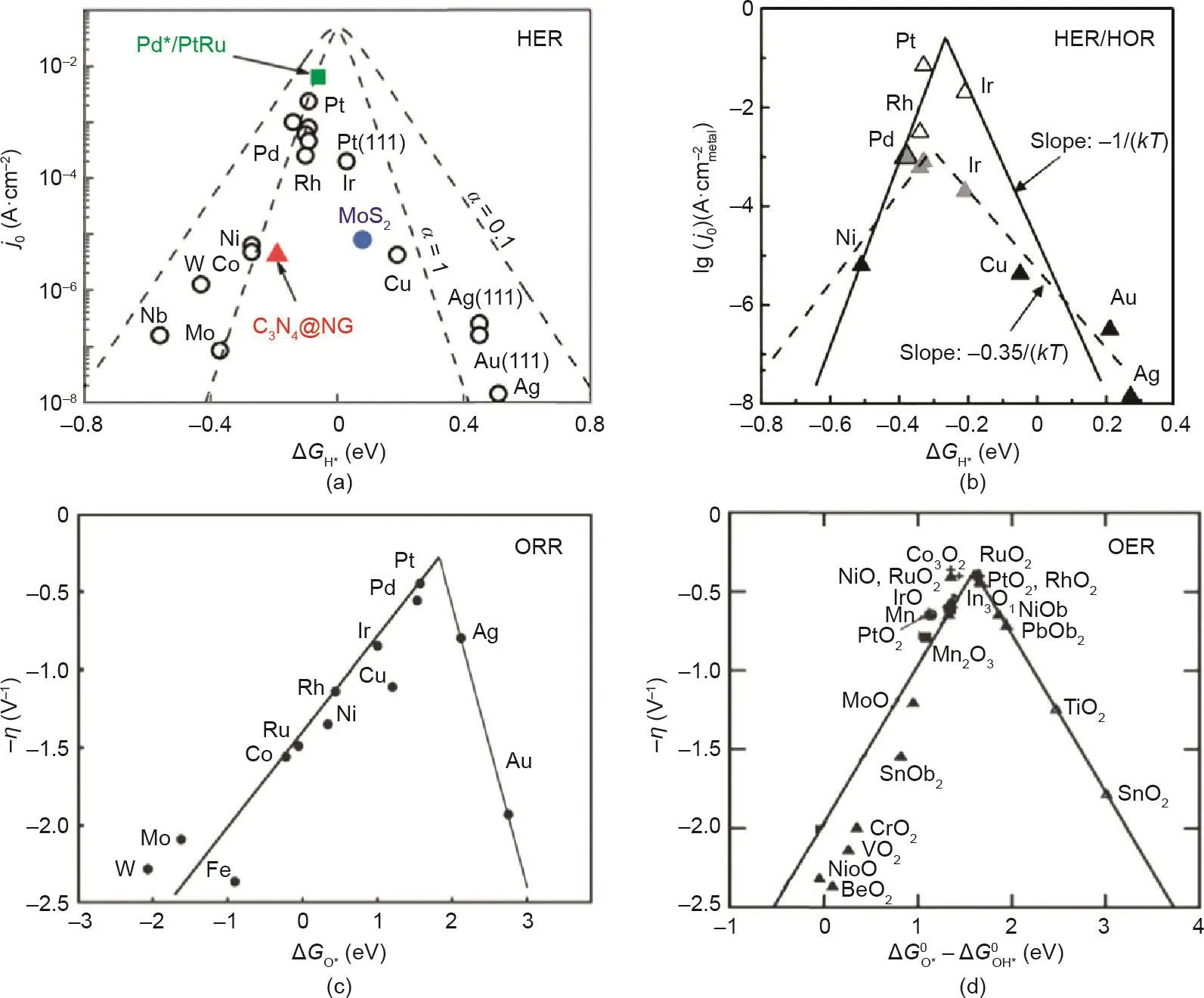
图4. (a)各种材料表面HER交换电流密度(j0)和ΔGH*的关系图[56];(b)酸性介质中表面归一化的HOR/HER交换电流密度(j0)对ΔGH*的火山曲线图[61];(c)不同金属的ORR活性和ΔGO*的关系图[63];(d)氧化物的OER活性(在规定电流密度下的过电位)对ΔGO*–ΔGOH*图[70]。
在燃料电池催化剂设计中,纳米结构的优化同样十分重要。在长时间的操作过程中,ORR催化剂中除了自身活性衰减外,还可能会因水淹而导致燃料电池快速失活[99,100]。由于多孔通道被积水阻塞,水淹将中断活性位点的氧气供应,导致淹没区域的ORR终止[101,102]。为了量化燃料电池中电催化剂的孔特性对其传质和抗水淹性能的影响,Wang等[103]为ORR催化剂设计了一种特殊的“拨浪鼓”状工作电极。双级孔隙Pt/C催化剂具有较大的孔容和规整的孔道结构,其传质性能和抗水淹性能是工业催化剂的4倍。事实上,不同类型的孔隙在ORR过程中具有特殊的作用。在ORR过程中,中孔和大孔对传质过程中更重要[104–106],而微孔有利于容纳大多数催化位点[107,108]。因此ORR催化剂需同时具有多级孔结构,以保证活性位点密度和传质效率。为构建催化剂多级孔结构,以二氧化硅胶体[105,109–111]、有序介孔二氧化硅[106,112–114]、聚苯乙烯微球[115]和其他一些氧化物[116,117]为模板的牺牲模板法得到了广泛研究和应用。例如,Liang等[118]使用胶体二氧化硅为模板合成了比表面积高达1280 m2·g–1的氮掺杂碳催化剂,且该催化剂为具有介孔/微孔分布的多级孔结构材料。然而,以上的牺牲模板法的模板去除步骤可能会十分耗时,且该步骤通常需要使用强酸或强碱溶液,这对研究人员和环境可能带来危害。为了避免这些缺点,Ding等开发了一种以NaCl重结晶为模板的形态学控制方法[119–122]。在这种方法中,NaCl模板可以用热水溶解而去除,且NaCl模板可循环使用。通过盐重结晶,具有特殊纳米结构的聚苯胺(PANI)被封装在NaCl晶体中,然后在高温下精确地转化为碳纳米材料(图6)。在高温焙烧过程中,碳纳米材料在NaCl晶体封闭的纳米反应器中气化,从而产生了大量的孔。所制备的3D-Fe/N-C催化剂具有多孔隙、高活性位点利用率的特点,对ORR具有良好的催化性能。
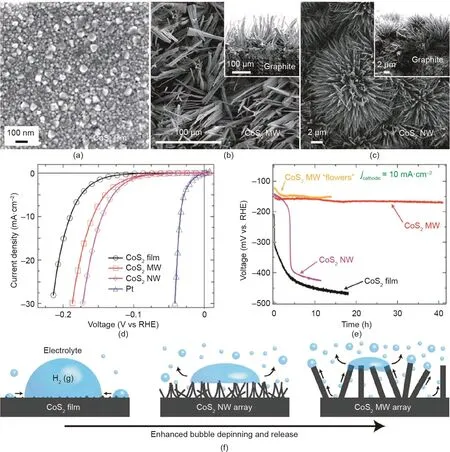
图5 .(a)~(c)不同形貌CoS2的扫描电镜图(SEM);CoS2电极电化学表征(d)和稳定性测试(e);(f)不同结构的CoS2氢气逸出示意图[92]。
3.2. 晶面工程
晶面调控是调节材料对给定反应的催化性能的一种广泛研究的方法。由于催化反应中间产物在催化剂不同晶面的吸附强度差异很大,催化材料的反应活性与其暴露晶面高度相关。晶面通常是用Miller指数表示的。纳米材料的暴露晶面与纳米颗粒的形状密切相关[123,124]。一般来说,纳米材料的晶面可分为低指数和高指数晶面类型[125]。低指数晶面是指Miller指数(hkl)三个组成部分之和较小的指数晶面,而高指数晶面至少包含一个大于1的Miller指数。
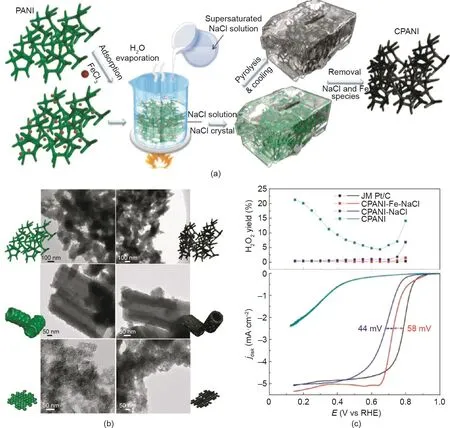
图6. (a)盐重结晶法形貌固定示意图;(b)所制的3D纳米结构聚苯胺(PANI)和对应碳化产物的透射电镜图(TEM);(c)不同样品的H2O2产出曲线和ORR曲线[119]。
低指数晶面纳米材料反应性的结构敏感性在单晶Pt催化析氢反应中已经得到证实。利用扫描隧道显微镜(STM,图7)证实了具有清晰表面的Pt (hkl)的表面形貌,并观察到其活性程度在碱性溶液中遵循(110)> (100) > (111)的顺序[126]。重要的是,Pt的不同晶面的活性趋势在碱性和酸性电解质中有很大的不同[127–129]。Markovic等[130]重点研究了Pt (111)和Pt岛修饰Pt (111),证实了这种pH效应涉及HER中的结构-功能关系。与原始Pt (111)表面的HER活性相比,Pt岛修饰Pt (111)电极上的HER活性在碱性溶液中提高了5~6倍,而在酸性电解液中的活性仅提高了1.5倍。pH值对其活性的影响被证明是由于边缘台阶位点解离水的特殊能力引起的[131–135]。此外,Pt (hkl)的活性顺序与低配位Pt原子的密度一致,这是由于低配位Pt原子加速了水的解离步骤[136–139]。在非吸附性HClO4电解质中,单晶Pt不同晶面的ORR活性也观察到类似的变化趋势[140]。当电解质替代为H2SO4时,Pt (100)比Pt (111)更为活跃[141–143]。这种活性差异是由于硫酸氢钠阴离子在Pt (111)上的特殊吸附行为所致。硫酸氢钠阴离子在Pt (111)表面的吸附比在Pt (100)表面的吸附更强,导致ORR的后续步骤受阻。以上研究证实了各晶面的不同性质对其催化性能有着显著的影响。在这项工作之后,许多研究集中在开发一种在催化表面上可控构建特定晶面的方法,以应用于理想的单晶金属及实用的纳米材料[144–150]。El Sayed等[144]首次发现了铂纳米晶体的晶面控制合成,制备得到富含(100)晶面的纳米立方体、(111)晶面的纳米四面体以及同时具有(111)和(100)晶面的纳米球。Sun等[145]报道了一种高温有机相法用于合成单分散(100)端Pt纳米立方体。晶面控制的Pt纳米立方体在酸性电解液中的ORR活性是工业Pt催化剂活性的两倍以上。
由于低配位原子、台阶、边缘和扭结的密度更高,具有高指数晶面的金属及化合物与典型的低指数材料相比通常具有更高的反应性[151]。然而,由于其较高的表面能,高指数晶面是热力学上不稳定的[152,153]。因此,制备高指数晶面纳米材料是一项艰巨的任务。近年来,为了提高催化活性,人们开发了多种方法来合成具有高指数晶面的纳米材料。Xia等[154]开发了一种在水溶液中还原的简单路线,以制备由(510)、(720)和(830)高指数晶面包围的Pt凹面纳米立方体(c-NCs)。利用甘氨酸控制H2PtCl6的还原动力学也可以制备得到Pt c-NCs[155]。Sun等[156]采用电化学方法合成了具有(730)、(210)和(520)晶面的四面体(THH) Pt纳米晶。除了单金属材料外,多金属高指数晶面纳米晶体的合成方法也得到了开发[157–160]。如图8所示,Guo等[159]报道了一类具有稳定高指数晶面和表面富Pt结构的新型Pt3Fe锯齿状纳米线(z-NWs)。这些独特的结构特征赋予了Pt3Fe z-NWs优异的ORR活性,其在0.9 V (vs. RHE)下的质量活性和比活性分别为2.11 A·mg–1和4.34 mA·cm–2。
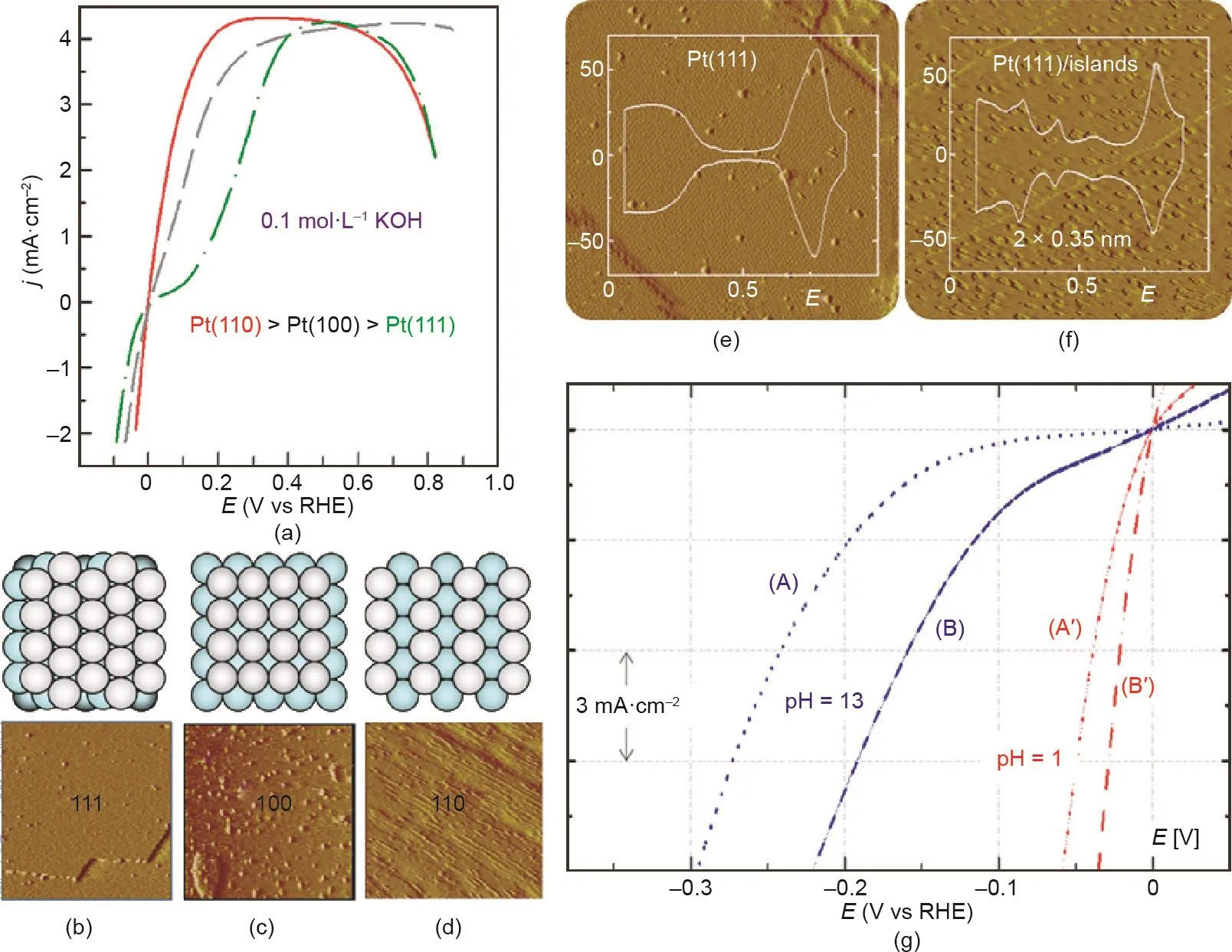
图7. (a)Pt不同晶面的HER/HOR;(b)~(d)Pt不同晶面的STM(插图为其对应的结构模型)[126];(e)制备的Pt (111)和岛状Pt/Pt (111)表面的STM图;(g)两个Pt晶面的HER活性曲线图[130]。
为了降低催化剂成本,设计和制备具有不同晶面的非贵金属纳米材料已成为晶面工程的研究热点[161–167]。Su等[168]研究了NiO晶体的生长机制,发现NiO晶体的表面能遵循(100) < (113) < (101) ≈ (110)的顺序。Han等[161]提供了一种无模板水热方法,用于可控地制备具有(001)、(112)和(001) + (111)晶面的Co3O4纳米立方体(NC)、纳米八面体(NTO)和纳米多面体(NP)。不同的晶面赋予Co3O4纳米晶表面的Co2+和Co3+活性位以不同原子结构。在含有丰富Co3+位点的还原氧化石墨烯(rGO)上,(112)晶面覆盖的Co3O4纳米颗粒对OER和ORR均表现出良好的活性。除了金属氧化物外,许多具有特殊晶面的金属化合物也相继被报道。Feng等[163]合成了具有稳定(210)面的Ni3S2纳米片阵列,展现了高效稳定的HER和OER电催化性能。Pan等[162]制得了具有不同晶体结构的花状磷化镍(Ni5P4和Ni2P),并证明其优异的催化活性归因于具有高能(001)晶面的多级结构。
3.3. 晶相工程
除了调整纳米晶体的暴露晶面外,调节原子尺度的排列(即晶相的转变)也会使催化剂的物理和化学性质发生根本性的变化,从而影响催化剂的本征活性。具有独特多晶相的过渡金属二硫化物是被广泛研究的典型案例。在这些多晶型中,亚稳态1T相由于其金属性的性质引起了极大的研究兴趣[169–174]。Jin等[169]用锂插层法从半导体2H-MoS2合成了金属性1T-MoS2纳米片。与相应的2H晶相相比,1T-MoS2在电催化HER方面表现出显著改善的性能(图9)。类似地,Jin等[173]通过更简单的微波辅助插层法合成了金属性1T二硫化钨(1T-WS2)。晶相工程赋予了1T-WS2更优异的导电性和更密集的活性位点,增强了其析氢催化活性。1T催化剂不仅可以通过改变原子排列获得独特的性质,而且其活性中心也可能不同于传统的2H结构相。Voiry等[175]通过去除化学剥落的MoS2纳米片表面的剩余负电荷,获得了具有优异活性的高导电性1T-MoS2纳米片。有趣的是,1T-MoS2和2H-MoS2部分氧化后,其活性变化形成鲜明对比。边缘氧化后1T-MoS2的HER活性几乎没有变化,但2H-MoS2的活性却严重下降。众所周知,通常2H-MoS2晶体的边缘是其主要的活性中心。部分氧化的1T-MoS2和2H-MoS2在HER活性上的显著差异表明,1T-MoS2催化的主要活性中心不是纳米片的边缘,而是纳米片的基面。
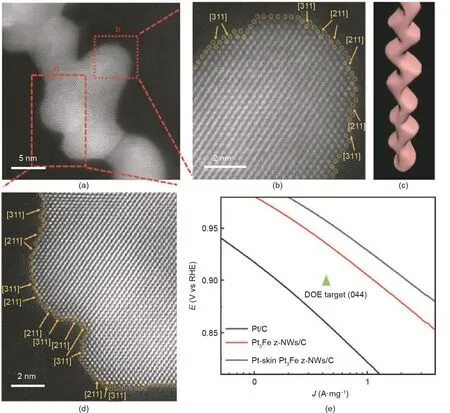
图8 .(a)Pt-skin Pt3Fe z-NWs的高角度环形暗场(HAADF)-TEM图;(b)、(d)图(a)中红色方块区域的放大图;(c)锯齿状纳米线的结构示意图;(e)商业化Pt/C、Pt3Fe z-NWs/C和Pt-skin Pt3Fe z-NWs/C的ORR质量活性图[159]。
金属氧化物的催化性能也受其晶相影响。Wu等[176–178]发现反转尖晶石晶体结构对尖晶石的ORR催化活性有很大影响(图10)。通过调整铁(Fe)的含量,Co–Fe基晶体的尖晶石结构可以从正常结构变为反结构,后再变回正常结构。电化学结果表明,具有反尖晶石结构的{Co}[FeCo]O4/NG具有最好的ORR活性。DFT结果进一步揭示了反尖晶石结构的{Co}[FeCo]O4/NG的高ORR活性是由于八面体位置的Fe和Co原子的异化效应引起的氧吸附能的改变和氧-氧键的拉长所致。此外,晶相对ORR反应途径的影响也有所报道。Karunagaran等[179]合成了四种不同晶相的氧化铁纳米颗粒负载于三维rGO气凝胶,并测定了它们催化ORR的电化学性能和电子转移。结果表明,在高电位(0.70 V)下,四种催化剂均通过双电子途径催化ORR。当电位降低到0.20 V时,ORR在含磁铁矿、磁赤铁矿和针铁矿的rGO复合材料通过四电子转移动力学进行,而在含赤铁矿的复合材料则通过两电子转移动力学进行。
过渡金属化合物的Jahn-Teller效应引起的构型畸变与其电催化性能的关系也有大量研究[180]。Liu等[181]通过对Co3S4/TETA杂化前驱体的超声剥离得到原子层厚度的Co3S4纳米片(CSATNs),并观察到CSATNs产物存在明显的结构畸变。CSATNs的结构畸变引起电子结构的改变。与块体样品相比[图11(a)、(b)],CSATNs的谱图向低磁场的偏移意味着其八面体中心()中Co3+的自旋状态从低自旋调整到高自旋。高角度环形暗场(HAADF)图像显示,八面体配位阳离子仅暴露在平面中,进一步揭示了Jahn-Teller拉伸的存在[图11(c)~(f)]。由于原子和电子结构的协同调整,CSATNs具有明显的增强OER性能。事实上,Jahn-Teller效应是由于简并轨道(t2g或eg)中心离子的电子分布不均匀造成的。因此,eg轨道上电子的填充态对过渡金属化合物的催化性能有着重要的影响。Shao-Horn等[182]进一步发现了钙钛矿基氧化物(ABO3)中B离子的eg轨道的填充状态与ORR活性之间的火山关系[图11(g)]。当钙钛矿型氧化物eg轨道上只有一个电子填充时(定义为eg≈1),其具有最高的ORR活性,因为此时O2可以以最佳结合能吸附在B位。虽然尖晶石的ORR活性位不是四面体位而是八面体位,但eg占有率理论也可以进一步扩展应用于尖晶石氧化物的活性描述[图11(h)][183]。
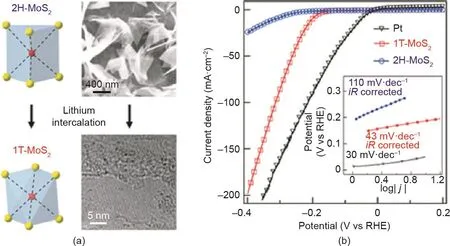
图9. (a)经过锂插层后由半导体2H-MX2到金属态1T-MX2的相转变;(b)两种形态MoS2的HER极化曲线[169]。
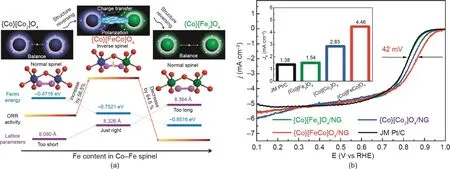
图10 .(a)结构转换和ORR活性之间的关系;(b){Co}[Fe2]O4/NG、{Co}[Co2]O4/NG、{Co}[FeCo]O4/NG和Pt/C的ORR活性[176]。
3.4. 非晶化
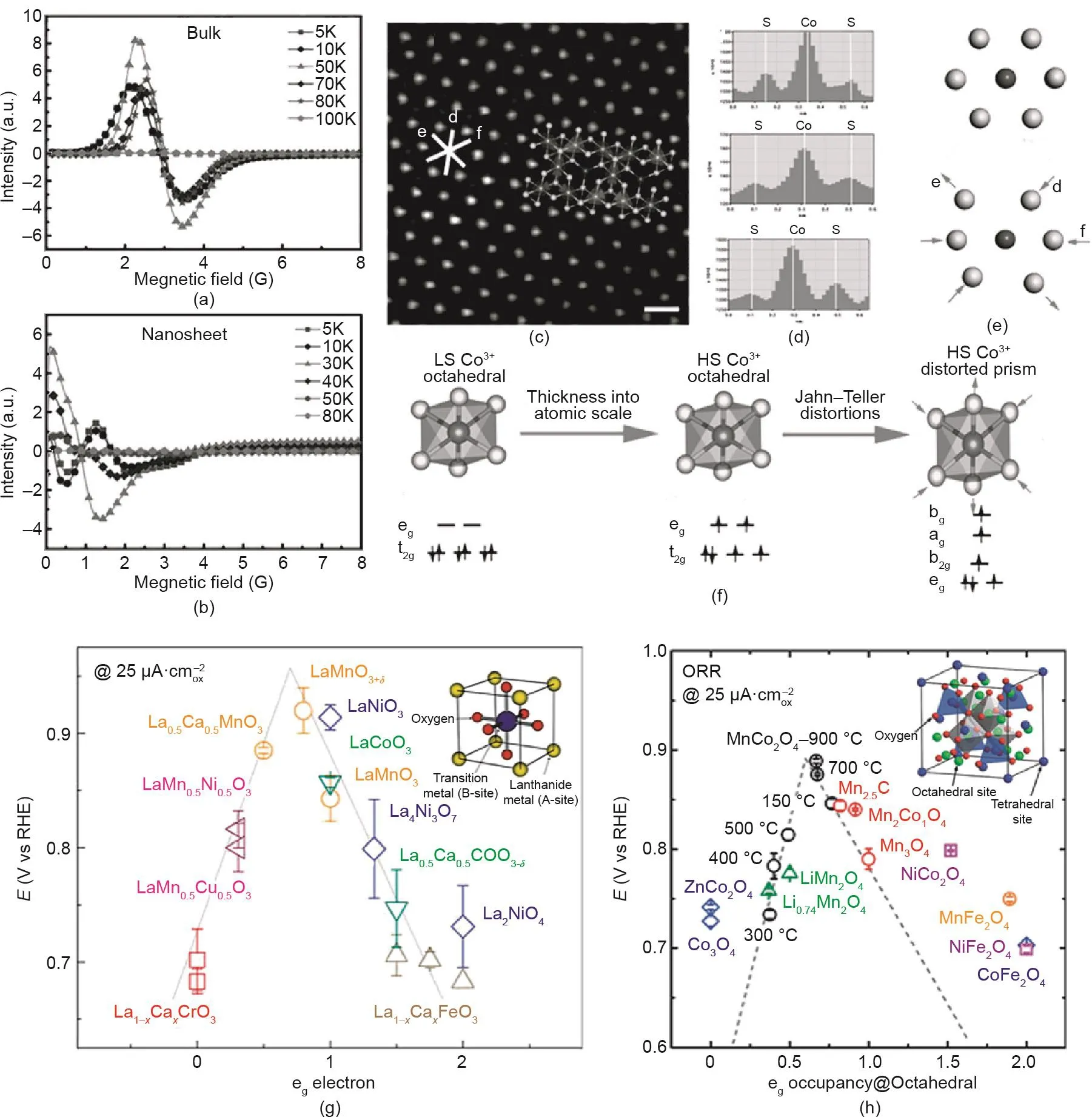
图11. (a)、(b)块状和Co3S4纳米片的电子顺磁共振图(EPR);CSATNs的HAADF(c)和强度线(d);Jahn-Teller变形(e)和结构转变(f)图示[181];(g)钙钛矿氧化物的ORR活性对eg电子的函数图[182];(h)在尖晶石氧化物的ORR活性中位于八面体位点的活性元素eg的作用[183]。
通过非晶化来调节原子尺度的排列,是提高材料催化性能另一个研究热点[184–189]。非晶相的短程原子排列有利于提高活性中心的密度[190–194]。早在1995年,Weber等[190]研究了非晶态化合物MoS3的结构单元,发现所有钼都处于Mo4+氧化状态,而硫原子则以两种不同的配位形式存在:S2–和S22–。Hu等[191,192]证实非晶态MoS2在催化HER方面更为活跃。非晶态MoSx薄膜表面极为粗糙,硫元素含量丰富,催化活性区大,活性中心密集。Benck等[192]进一步揭示了非晶态硫化钼的HER活性的增加是非晶态结构和纳米结构所引起的大量活性位点的作用。同时,Li等[195,196]从组成和结晶度方面系统地研究了非晶态MoS2催化活性的来源。有趣的是,实验结果表明结晶度是决定催化性能的关键,而组成并不特别重要。
除了HER催化,Smith等[185]基于对铁、镍和钴混合金属氧化物的研究,证明非晶态材料比晶体材料具有更佳的OER催化活性。对于非晶态结构,金属在整体材料中的分布是均匀的,且其成分可以精确控制。以最佳元素含量配比制备的a-Fe100-y-zCoyNizOx催化剂具有优异的催化性能,甚至可以与工业贵金属氧化物催化剂相媲美。基于非晶态材料的组成可控的特点,可以进一步研究金属组成以及非晶化对电催化性能的影响。Smith等[184]制备了21个复合金属氧化物薄膜用于电催化水氧化,并测量了每个样品中Fe、Co和Ni的准确化学计量浓度。电化学测量证实铁含量对降低Tafel斜率很重要,而钴或镍有利于降低过电位(图12)。由于无定形态催化剂优异的催化性能,其规模化生产方法的研究十分重要。Kuai等[186]提出了一种喷雾辅助的方法,通过这种方法可以可持续地获得非晶混合金属氧化物,非常适合工业应用。所制备的Fe6Ni10Ox在电化学OER中驱动10 mA·cm–2时表现出0.286 V的低过电位,且Tafel斜率仅为48 mV·decade–1,优于所有研究的Fe-Ni-Ox催化剂的催化性能。
虽然非晶态工程可以大大提高催化剂的活性位密度,但由于晶体结构的短程无序,非晶态材料的导电性会降低。将低导电非晶态材料与高导电材料复合是保证非晶催化剂优良电催化性能的有效途径。Lee等[197]合成了以低成本、高导电性的Ketjenback(KB)碳负载非晶MnOx纳米线作为高效ORR电极,大大加速了电催化过程中的电子转移。许多其他非晶态/导电复合材料,如非晶态MoSx/碳复合催化剂[198]、非晶态MoSx/聚吡咯共聚物薄膜(PPy/MoSx)[199]和非晶态MoSx/氮掺杂碳纳米管催化剂(NCNT)[200]也被报道。这些复合材料中的高导电骨架可以克服非晶态催化剂的低导电性引起的障碍,从而显著提高催化活性(图13)。多孔金属纳米结构,如镍泡沫[201]和纳米多孔金[202],也被用作支持非晶态MoSx催化剂的导电基底,以显著增强其HER活性。
3.5. 缺陷工程
缺陷普遍存在于纳米材料中。人们已经认识到,缺陷催化剂表面总是比无缺陷催化剂表现出更高的反应活性[203–206]。因此,缺陷工程逐渐发展成为调整纳米材料电子和表面性质的有效方法[207–209]。Cheng等[210]通过在水溶液中还原非晶态二氧化锰,合成了四方或立方MxMn3–xO4尖晶石。纳米MxMn3–xO4由于其高活性面积和丰富的缺陷,对ORR和OER都具有相当高的催化活性。同样,Qiao等[211]合成了富氧空位(OV)缺陷的介孔MnCo2O4材料,发现其稳定性和耐甲醇性能甚至超过了Pt/C催化剂。为了深入了解缺陷对催化性能的影响,Li等[212]应用DFT+U计算研究了OV浓度对β-MnO2催化剂电子结构及其ORR的催化性能的影响。如图14所示,电子结构与OV浓度的曲线关系表明,OV浓度可以调节β-MnO2的电导率和ORR催化活性。适当浓度的OVs将大大提高MnO2的电导率,而过量的OVs将阻碍ORR过程。缺陷工程也可用于提高纳米材料催化活性位点的密度[203,213,214]。Xie等[203]设计了一种高浓度前驱体、不同硫脲用量的反应,实现了对所制备超薄MoS2纳米片缺陷的可控调制。由于其富含缺陷的结构,MoS2纳米片表面形成了许多细小的裂纹,使得其活性位点数量是无缺陷MoS2的13倍。
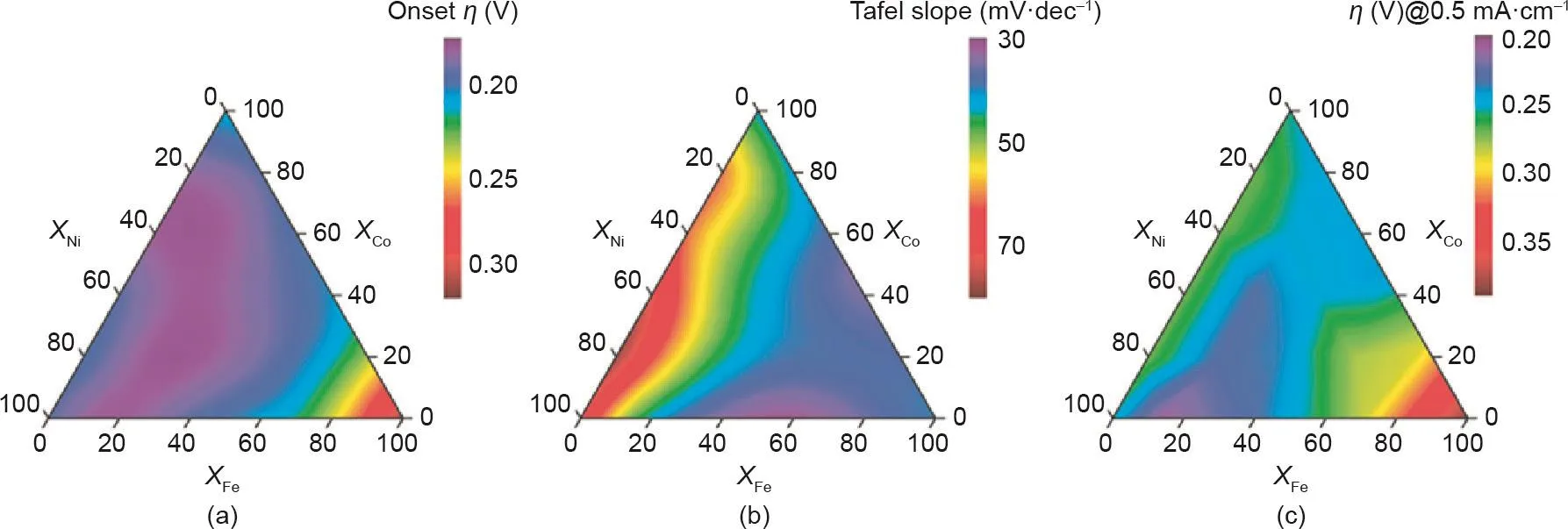
图12. 不同金属成分的无定形金属氧化物薄膜活性参数的等高线。(a)起始电位;(b)塔菲尔曲线;(c)j = 0.5 mA·cm–2时的过电位[184]。
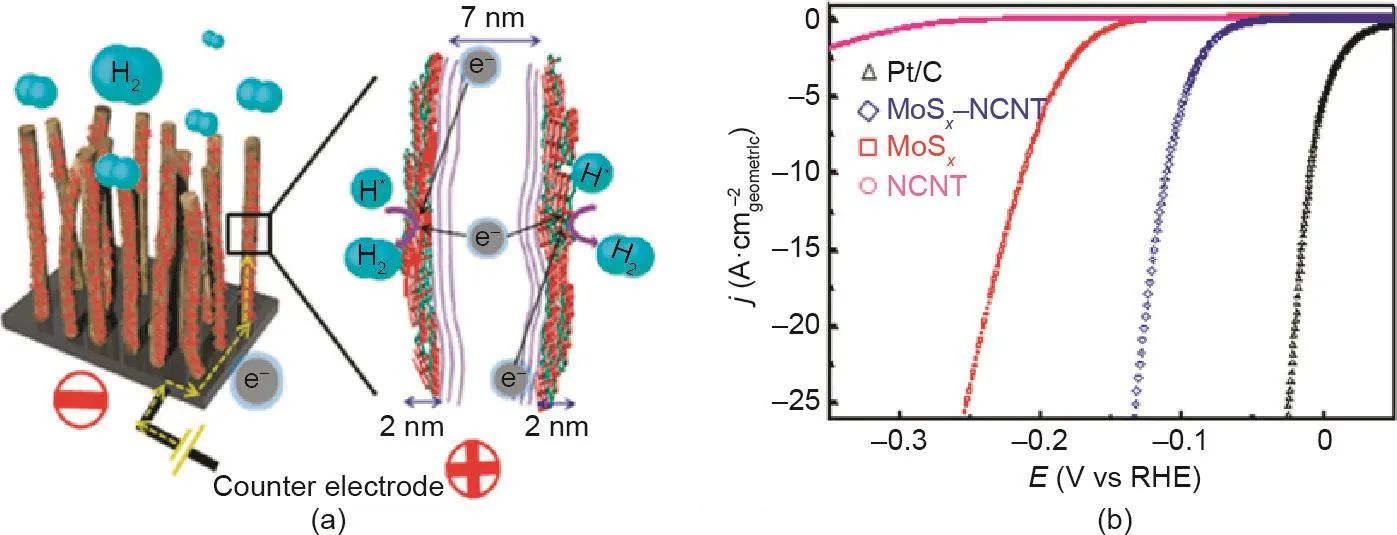
图13. (a)MoSx/NCNT森林状杂化催化剂的HER示意图;(b)不同样品的HER活性曲线[200]。
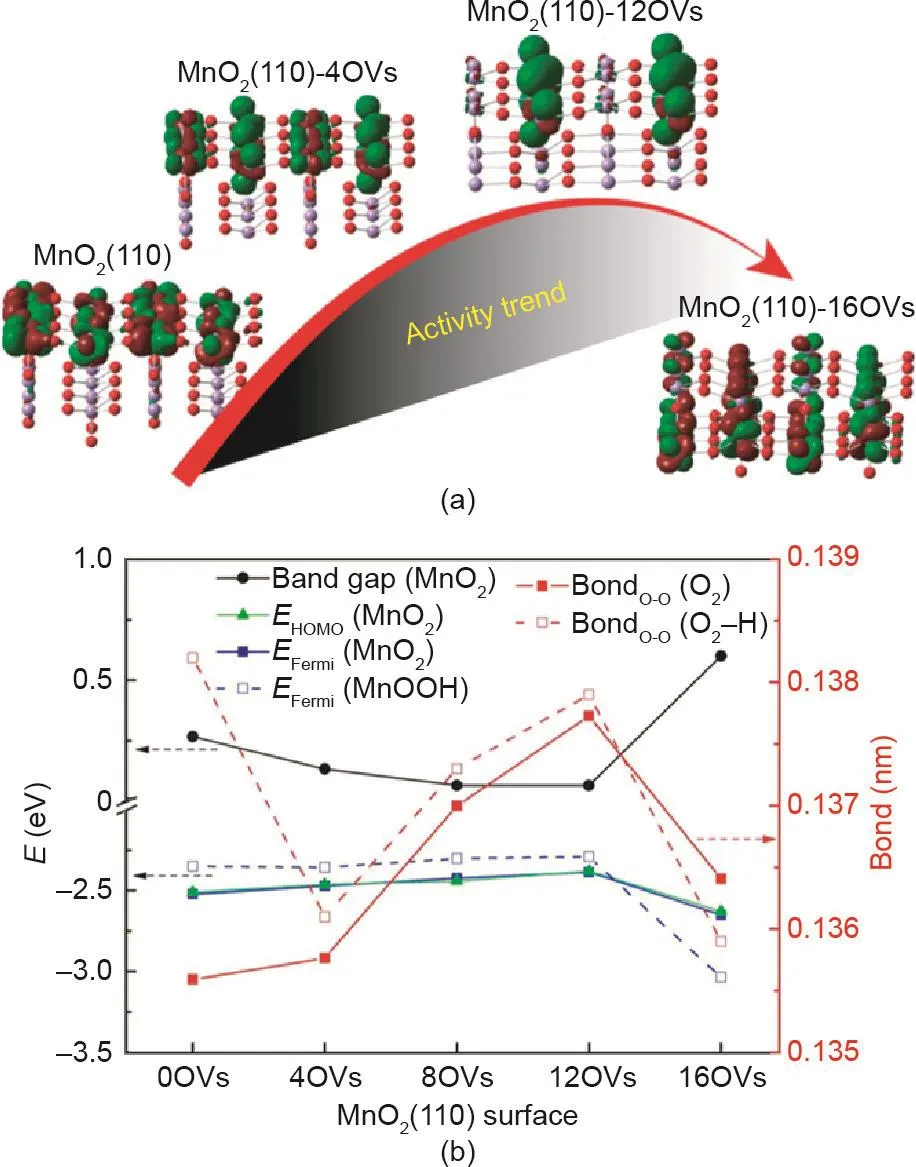
图14. (a)不同OV浓度下MnO2的活性趋势;(b)DFT计算各性质结果随OV浓度的改变[212]。
与金属化合物中的空位类似,碳基电催化剂中的固有缺陷是普遍存在的,但长期以来却一直被忽略[209]。通常,碳材料经过杂原子掺杂后容易形成缺陷,成为有利于电催化的活性中心[215,216]。然而,杂原子掺杂碳基材料的电催化活性主要归因于杂原子掺杂的诱导效应。随着时间的推移,一些研究发现,具有本征缺陷的纯碳催化剂的催化活性甚至优于杂原子掺杂碳材料[217,218]。Hu等[219]发现缺陷碳纳米笼(CNC)具有比硼掺杂碳纳米管更高的ORR活性。Hu等成功地合成了缺陷丰富的CNC,该材料具有许多典型的缺陷位置,但没有任何掺杂剂[图15(a)]。电化学结果表明,缺陷密度最高的CNC材料表现出最好的ORR电化学活性[图15(c)]。DFT结果进一步表明,这些缺陷材料的高ORR活性可归因于五边形和锯齿形边缘缺陷[图15(d)]。Yao等[220]利用第一性原理计算,预测了石墨烯上585种缺陷的ORR活性甚至超过了氮掺杂位点,并通过实验研究为这一理论预测提供有力支持。考虑到缺陷机制,Yao等[221]通过在950 ℃下碳化Zn-MOF制备了一种无元素掺杂的多孔碳(PC)材料。通过去除锌原子,可以在PC催化剂上形成缺陷,使PC催化剂不仅具有优异的ORR活性,而且具有与商业Pt/C催化剂相当的稳定性。此外,除ORR过程外,在从氢-水转化系统中其他三种电化学反应(即HOR、OER和HER)的电催化活性被证明对石墨烯中的缺陷类型特别敏感[222]。
3.6. 原子掺杂
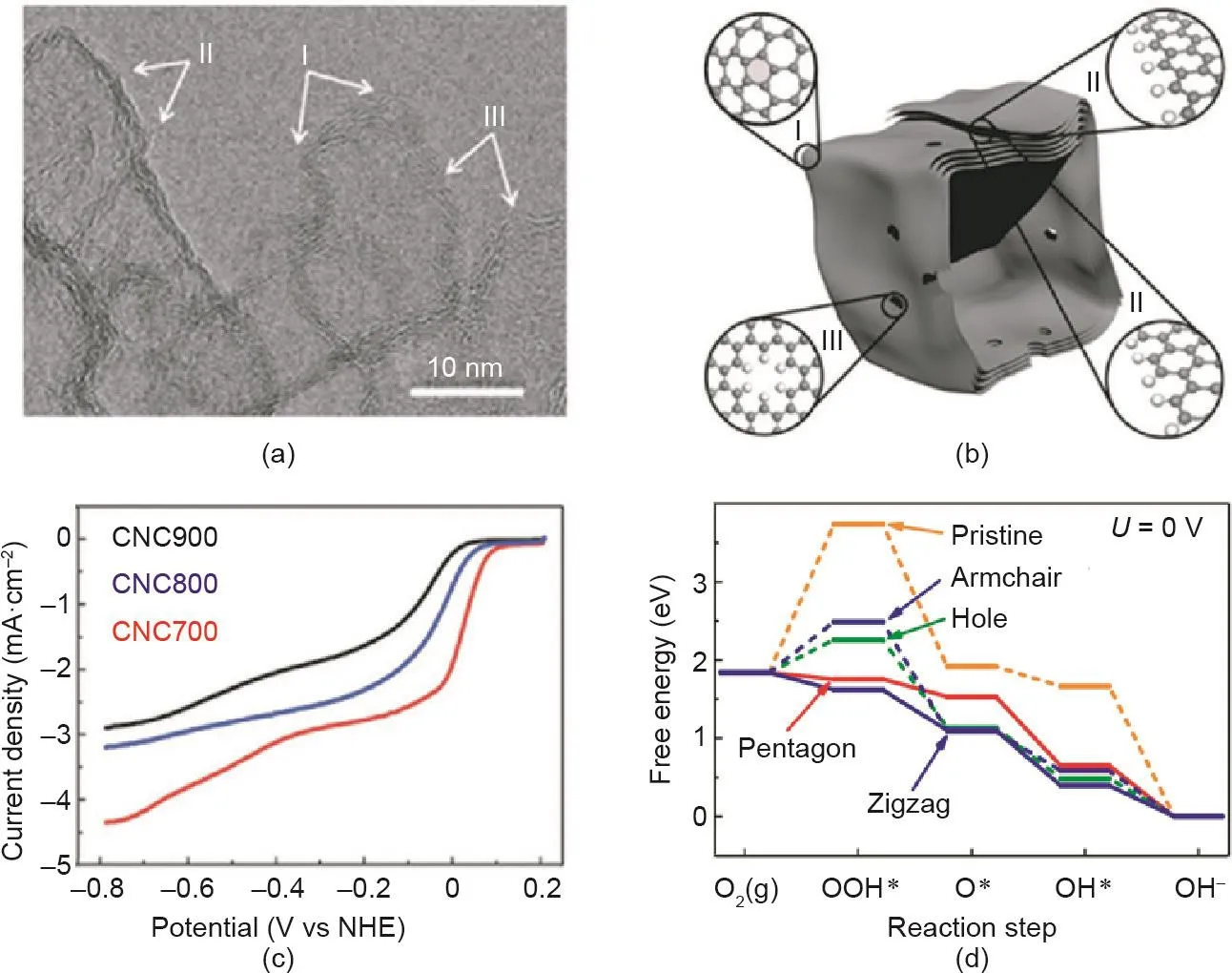
图15. CNC700的高倍透射电镜图(a)和结构示意图(b);(c)CNC样品的旋转圆盘电极(RDE)ORR活性曲线;(d)不同缺陷的ORR步骤的自由能图[219]。
原子掺杂是调节催化材料性能最常用的策略。通过合理地将一个或多个金属或非金属元素引入材料的晶格中,可以调节原材料的电子结构,从而有效地提高材料的催化性能[223–232]。以MoS2为例,多种金属元素如Ni、Co、Fe、V、Li和Cu被成功掺杂到其晶体结构中,有效改善了其物理和化学特性[171,233–237]。在这些掺杂的金属元素中,Ni和Co倾向于掺杂在MoS2中S元素的附近,这将降低S边缘的氢吸附能,增加MoS2材料中活性位点的密度[233–235]。与Ni和Co的掺杂不同,V掺杂不能增加活性位点的数量,但会增强MoS2的导电性[236]。Xiong等[97]通过结合实验和理论计算,探讨了碳化钼中镍的掺杂对其表面电子结构的影响及与其催化性能之间的关系。如图16(a)~(d)所示,通过水热和碳化处理,一维NiMo2C纳米线阵列直接构建在导电泡沫镍上(NiMo2C/NF)。与Mo2C和Ni催化剂相比,这种无黏结剂的NiMo2C/NF集成电极显示出更优异的催化活性[图16(e)]。DFT计算结果表明,Ni与Mo2C晶格的结合改变了催化剂的电荷分布,产生了Ni与Mo2C的协同效应,从而降低了氢结合能[图16(f)、(g)]。除了金属元素,非金属元素掺杂的研究也非常活跃。Xie等[238]成功合成了氧掺杂MoS2超薄纳米片,可以协同调控其活性位点和导电性。DFT计算结果显示,含氧MoS2的微分结合能更小,其驱动HER过程的能垒更低。Zhang等[239]通过部分磷化金属氧化物前驱体,构建了氧掺杂NiMoP2(O-NiMoP2)。如图16(h)~(i)所示,氧的掺入优化了NiMoP2表面的氢吸附能,其ΔGH*比未掺杂样品更接近于零。此外,O-NiMoP2中的Ni和Mo带有更多的正电荷,这有利于水分子的吸附和活化,极大加速了水在碱性介质中的解离。
除了金属化合物,碳基材料也被广泛用作掺杂目标,极大地扩大了催化剂的研究范围[240–245]。在2013年年初,Li等[246]报道了一种磷掺杂石墨烯,其ORR催化性能与商业用Pt/C相当。随后,Li等[247]进一步制备了氮磷双掺杂石墨烯材料作为ORR和OER的电催化剂,其催化活性超过了商业Pt/C催化剂。为了揭示杂原子掺杂碳高活性的起因,Yang等[248]对一系列不同杂原子掺杂的石墨烯进行了全面的DFT计算。DFT结果表明,掺杂碳催化剂存在三重效应,即电荷、自旋密度和配体效应决定了掺杂碳催化剂的本征催化活性及ORR机制(图17)。当碳材料被单一杂原子掺杂时,掺杂原子周围的碳位只能被单一效应激活。这导致ORR通过结合机制进行,并且存在不低于0.44 V本征过电位限制。当碳材料被金属或双杂原子掺杂时,双碳位可以被三重效应激活,其ORR遵循离解机制。此时,结合机制的活性限制将不再有效,从而ORR活性可得到有效增强。Huang等[249]合成了金属和非金属元素共掺杂的石墨烯,并揭示了氮掺杂石墨烯中的氮结构和微量金属原子对HER催化的作用。该研究发现,季铵态氮是氮掺杂石墨烯中三种掺杂氮类型中最活跃的位点。而当掺杂微量钴原子时,平面(吡啶和吡咯)氮的活性最强;当微量钴原子被镍取代时,平面(吡啶和吡咯)氮的活性则被抑制。
3.7. 界面工程
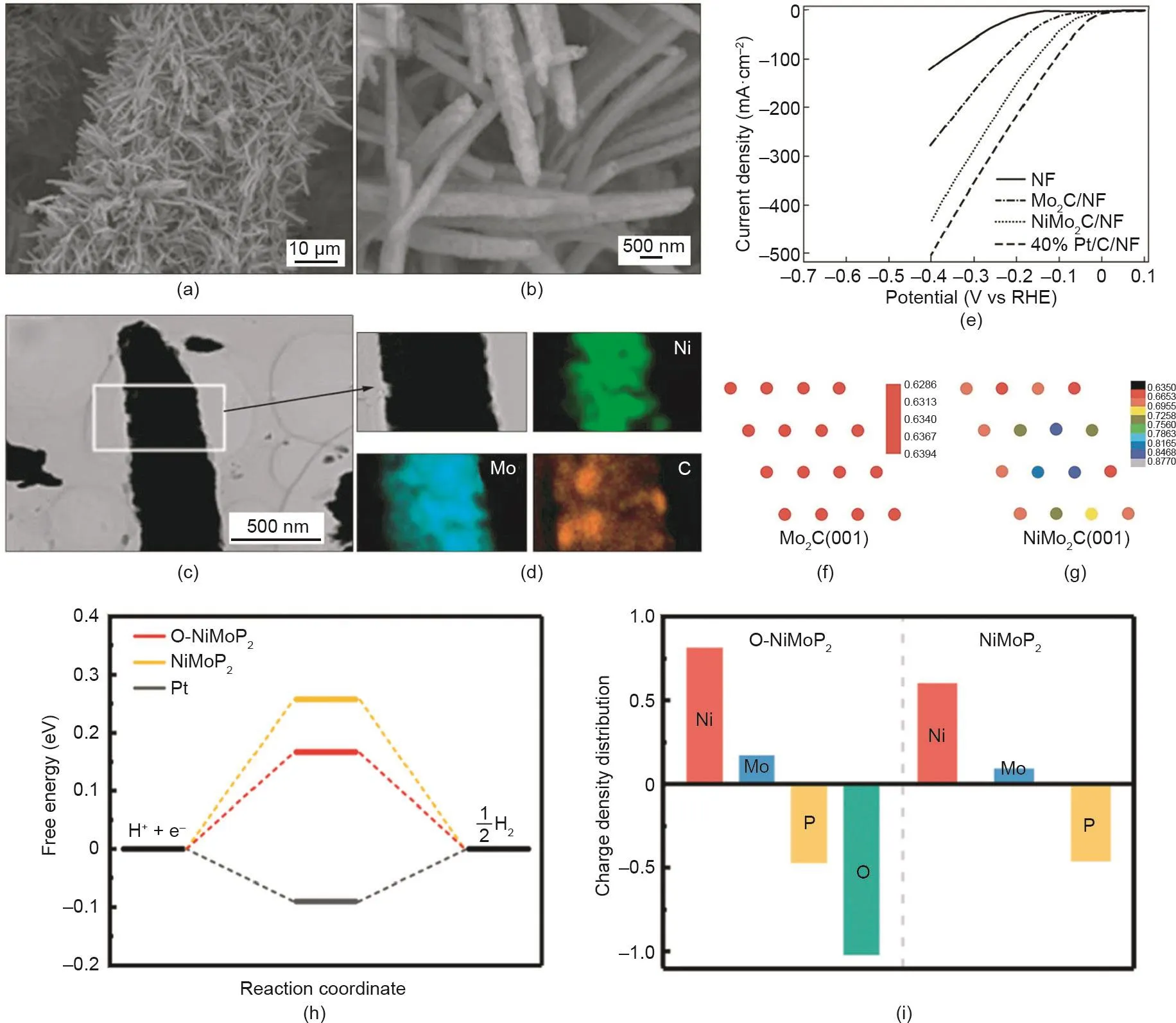
图16. (a)~(d)NiMo2C电极的扫描电镜、透射电镜图和元素能谱图;(e)NiMo2C电极的电化学析氢活性曲线;Mo2C (001)(f)和NiMo2C (001)(g)的Bader电荷分布图[97];不同NiMo基样品的氢自由能(h)和电荷密度分布(i)图[239]。
杂化纳米材料拥有一个位于两个组分的边界的界面[250]。对于多相催化剂来说,具有适当的界面结构是极其重要的,因为界面区域总是呈现出独特的物理和化学性质[251]。这些独特的性质可以促进材料结合、转化和运输表面物种(如吸附物种、电子和中间体)的能力,极大促进了发生在其表面的催化反应[252–255]。近年来,有大量的研究通过界面工程设计与合成催化剂用于氢-水转化。一般来说,根据组分的相对位置,杂化材料可分为支撑结构、异质结构或核壳结构[256]。支撑结构的特点是支撑组分比其他组分大得多,而异质结构材料中的组分尺寸相似。在核壳结构中,一个组分被另一个组分覆盖,在两个组分之间的边界处存在界面。这三种具有不同界面结构的杂化材料是近年来研究金属、金属氧化物、非氧化物等组装的典型结构。例如,Feng等[257]通过控制镍原子在煅烧过程中向外扩散,合成了一种MoNi4固定在MoO2长方体上的支撑结构电催化剂(MoNi4/MoO2@Ni),在碱性溶液中表现出优异的HER活性[图18(a)、(b)]。Xue等[258]通过将金属钴逐渐磷化为CoP制备了异质结构Co/CoP纳米颗粒。通过改变NaH2PO2和Co元素的比重,异质Co/CoP纳米颗粒中的CoP含量能够可控调节,进而改变Co/CoP催化剂的界面区[图18(c)~(f)]。如图18(g)~(h)所示,Li等[259]通过对双金属(Ni, Co)有机骨架进行碳化还原和可控氧化煅烧,制备了由碳限域NiCo@NiCoO2纳米颗粒组成的多孔纳米阵列(NiCo@NiCoO2/C PMRAs)。所制得的NiCo@NiCoO2/C PMRAs包含了催化OER所需的几种特性,包括大的表面积、良好的导电性和丰富的电催化活性位点。
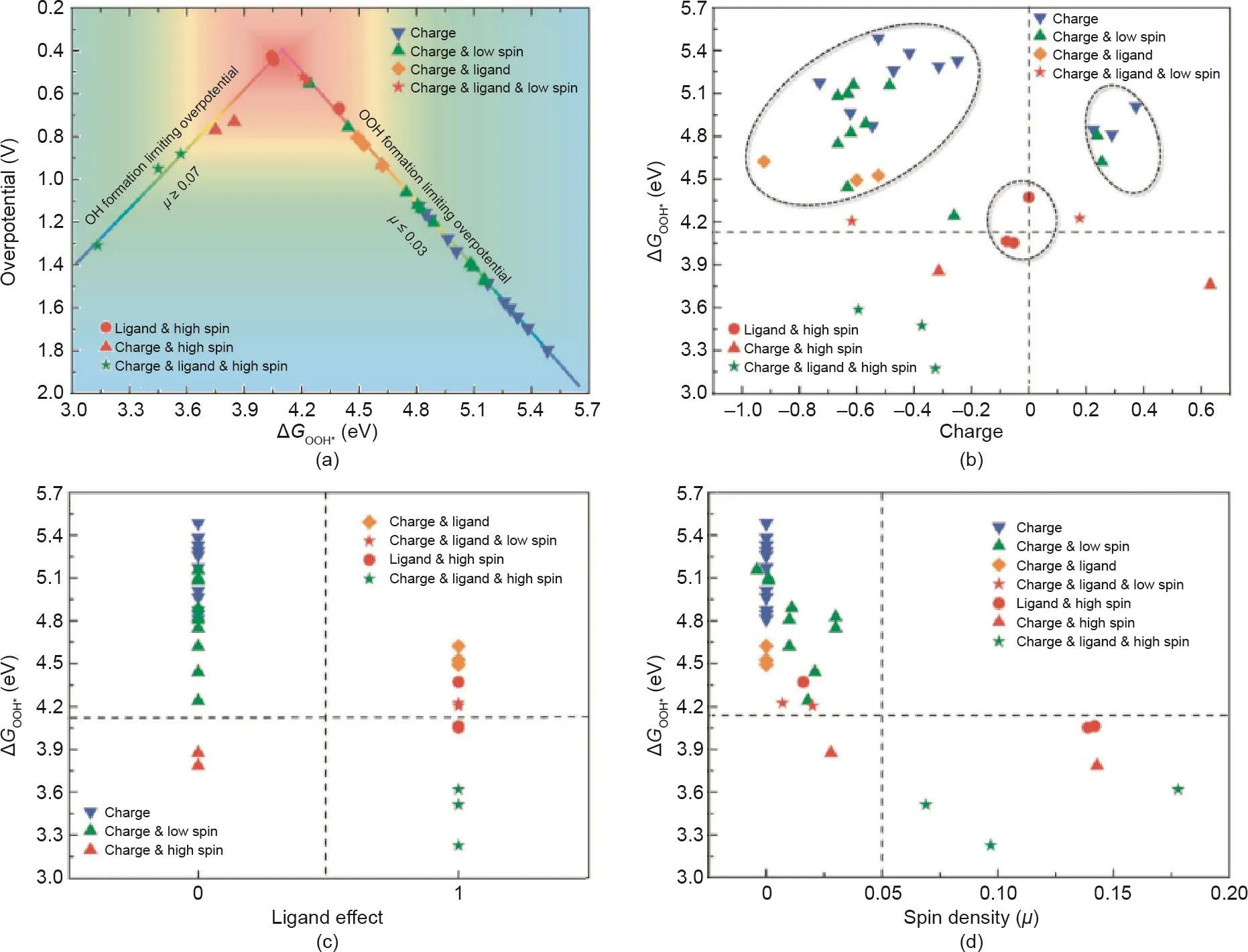
图17. (a)碳活性位点的ORR过电位对ΔGOOH*函数图;ΔGOOH*对电荷效应(b)、配体效应(c)和自旋密度关系(d)图[248]。
为了更合理地设计催化剂,对具有丰富界面的杂化材料的催化性能提升原因也进行了深入研究。其中,杂化材料中界面位点的电子结构调控已被充分证实[260–267]。Yu等[268]用X射线吸收近边光谱(XANES)观察到了Ni(OH)2/Pt催化剂的电子转移。在α-和β-Ni(OH)2/Pt电极上Ni的前边缘和主吸收边缘均向低能方向移动,说明电子从Pt基底向氢氧化物转移。Hu等[269]揭示了Pt/CoS2杂化体系中CoS2和Pt在水电解反应中的强金属-载体相互作用(SMSI)。DFT计算预测了Pt的d带结构向下移动,这也进一步被X射线光电子能谱(XPS)和X射线吸收精细结构(XAFS)所证实。尽管电子转移在金属/金属复合催化剂中已经得到了很好的证明,但其高催化活性的潜在机制尚不明确。为了揭开这个谜团,Peng等[270]研究了金属/金属氧化物界面的化学性质,发现了一种界面诱导的协同效应——“烟囱效应”[图19(a)、(b)]。DFT计算结果表明[图19(c)、(d)],界面附近的位点对H2O*和OH*物种不吸附,而只选择性地吸附H*,这有效地避免了H2O*和OH*对活性位点的毒害作用。同时,界面上的活性位上H*反应物种的ΔGH*接近于0,说明其对H*反应物种的吸附和解吸能力良好。结合以上特点,析氢反应在金属氧化物/金属催化剂界面处连续进行,形成了类似于连续产氢的烟囱。此外,金属氧化物/金属复合材料的HER活性与界面金属原子之间呈正相关[图19(e)],说明可以通过增加界面活性位点的数量从而加速析氢过程。除金属/金属复合催化剂外,Peng等还通过制备单金属NiONi3S2异质节纳米片进一步研究了金属/金属复合催化剂性能提高的根源[271]。NiO-Ni3S2界面处Ni–S键向Ni–O键的电子转移导致了其比基准Pt/C和RuO2催化剂具有更好的电解水活性[图19(g)]。DFT计算结果表明,NiO-Ni3S2界面上氢(或含氧)中间体的活化势垒显著降低,说明界面活性位点具有优异的OER和HER本征活性[图19(h)、(i)]。
空位形成是界面材料的催化性能提升的另一个重要因素[272–277]。Xi等[278]报道了一个典型FeS2/CoS2杂化纳米片(FeS2/CoS2-NSs)用于催化水电解。在合成过程中,通过采用共沉淀法制备了CoFe2O4纳米粒子,后通过硫化将其转化为FeS2和CoS2相,并产生包含缺陷位点的界面。电子顺磁共振(EPR)谱显示,FeS2/CoS2-NSs复合材料在g= 2.007处具有较强的EPR信号,这表明其具有丰富的S空位。扩展X射线吸收精细结构(EXAFS)进一步研究样品的局部结构,检测到FeS2/CoS2-NSs中Fe的K边EXAFS的峰强度明显降低,这说明了Fe的配位缺陷。Qu等[279]在具有嵌入式结构CeO2/NiO(Ce-NiO-E)或表面负载结构CeO2/NiO(Ce-NiO-L)发现了类似的现象。NiO(Ni3+:62%,氧缺陷:24%)、Ce-NiO-L(Ni3+:69%,氧缺陷:26%)和Ce-NiO-E(Ni3+:71%,氧缺陷:32%)中的Ni3+和氧缺陷的增加趋势与活性趋势密切相关,表明界面区的空位对活性的增强有很大贡献。事实上,界面材料的电子结构调控和空位并不独立存在,界面材料的催化性能可能同时受到这两个因素的影响。
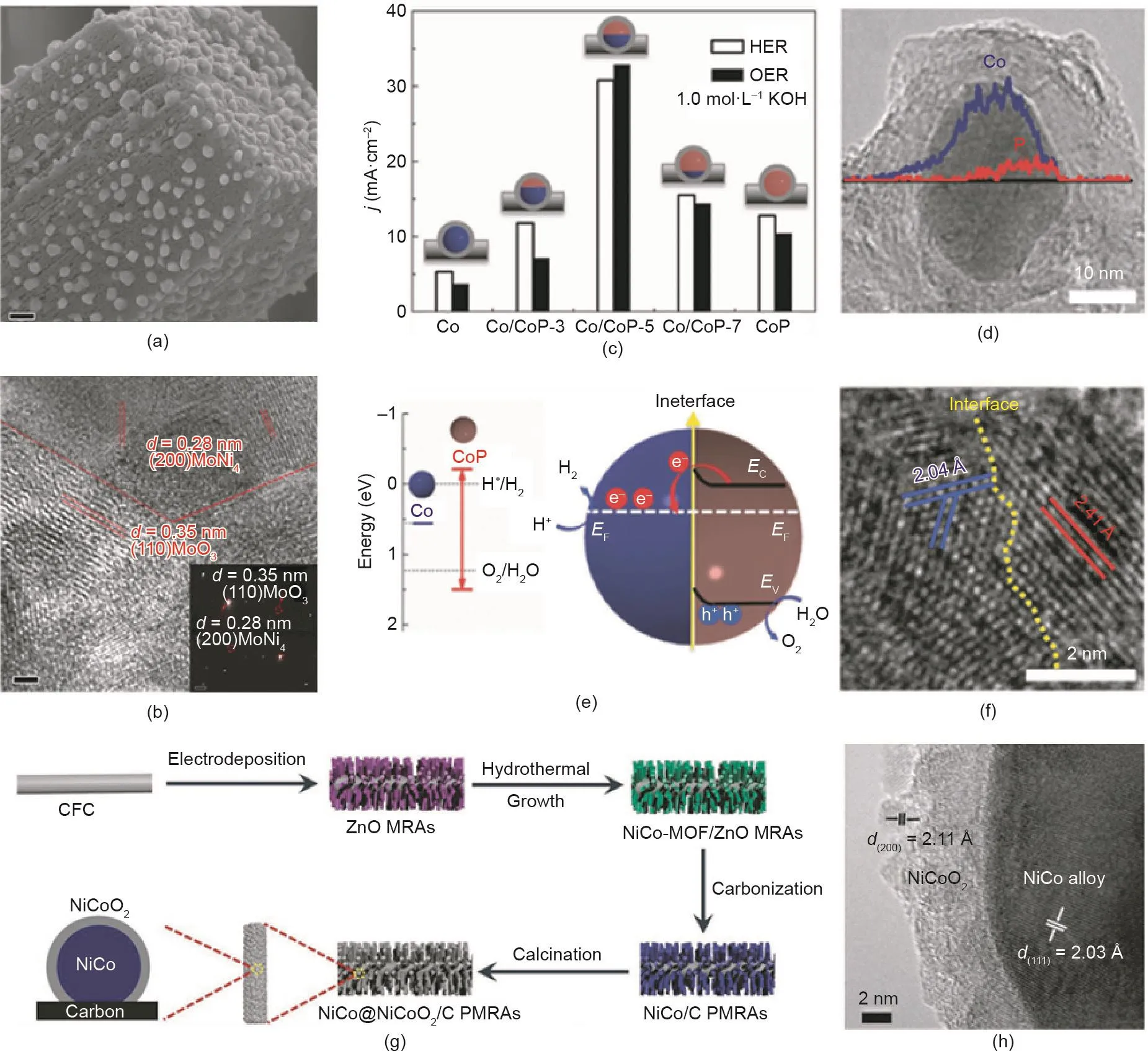
图18. (a)、(b)MoNi4/MoO2@Ni的典型SEM图[257];(c)制备样品的电流密度;(d)Co/CoP-5纳米颗粒的组成曲线;(e)金属Co、CoP和基于Co/CoP的Mott-Schottky接触的电子结构;(f)Co/CoP-5纳米颗粒的HRTEM图[258];NiCo@NiCoO2/C PMRAs的制备过程(g)和HRTEM图(h)。
3.8. 合金化
合金化是两种或两种以上金属的金属原子相互扩散渗透,或通过熔化、烧结或气相沉积过程将非金属元素添加到金属中。合金化是提高金属催化剂性能的有效策略,它不仅可以细化晶粒尺寸,提高机械强度以及催化剂的比表面积,还可选择性地减少单一组分(如Pt、Au、Ru等贵金属)的用量,以降低催化剂的成本。此外,由于组分之间的协同作用,通过加入其他元素形成合金,可以改变金属的催化活性和选择性[280–284]。根据Brewer-Engel价键理论[285],将具有未填充d轨道的金属和具有内部成对d电子的金属合金化可以调节合金表面的氢吸附能,从而提高析氢活性。Raj等[286]采用电沉积技术制备了一系列镍基二元复合材料,在碱性溶液中的HER催化活性变化趋势是:Ni-Mo>Ni-Zn>Ni-Co>Ni-W>Ni-Fe>Ni-Cr。Ni-Mo合金因其优异的催化活性被认为是最有前途的HER催化材料。Zhang等[287]利用磁控溅射技术在镍基表面成功地构建了一层尺寸均匀、元素分布均匀的Ni-Mo合金纳米棒,并发现其交换电流密度几乎是单金属催化剂的10倍。研究表明,Ni-Mo合金电极优异的催化活性主要来自两个方面:①在生长过程中,双组分金属的晶粒细化导致比表面积增加;②元素Ni和Mo的电负性差异导致电子在Mo周围聚集,从而形成电催化的协同作用。
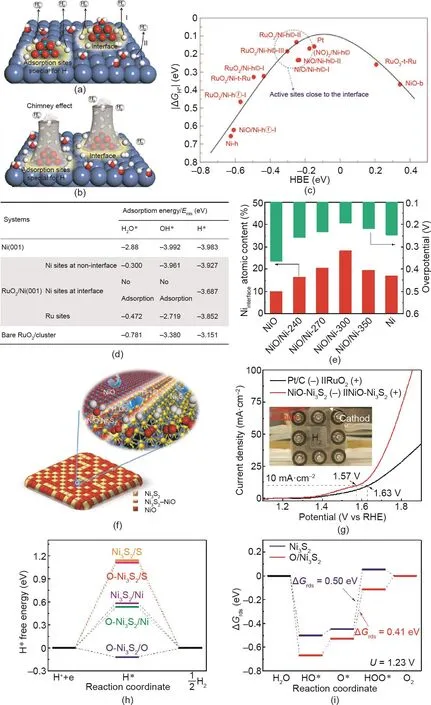
图19. (a)RuO2/Ni复合催化剂中两种可能的HER路径图示;(b)在金属/金属氧化物界面处的“烟囱效应”;(c)ΔGH*对氢吸附自由能的曲线;(d)不同团簇上H2O*、OH*和H*物种的吸附能;(e)NiO/Ni样品HER活性随Niinterface含量的改变趋势[270];(f)单金属NiO-Ni3S2杂化纳米片的水裂解图示;(g)NiO-Ni3S2及Pt/C和RuO2电极组合的水裂解极化曲线;Ni3S2和O-Ni3S2表面的氢吸附自由能(h)和OER各步的反应自由能图(i)[271]。
为了减少贵金属的使用,许多非贵金属(即Co、Ni、Fe、Cu、V、Cr、Mn、Zn等)被用来与贵金属结合形成合金作为电催化剂[206,288]。对不同PtM合金的ORR性能研究表明,其活性顺序为:PtFe/C>PtCo/C>PtV/C>PtNi/C>Pt/C [289–291],稳定性趋势[292]为:Pt3Ir (111) >Pt3Co (111) >Pt3Ni (111) >Pt3Fe (111)。此外,Stamenkovic等[293]根据铂合金的ORR活性与3d金属的d带中心位置,发现了典型的火山关系(图20)。研究表明,Pt3M催化剂的ORR机制是O2解离或质子/电子转移到O2分子上,最佳ORR催化剂的氧结合能应比Pt弱。Bampos等[294]在酸性溶液中进一步合成了一系列碳负载Pd-M(其中M=Ag、Co、Cu、Fe、Ni或Zn)双金属催化剂,其活性变化趋势为:PdZn/C>PdNi/C>Pt/C>Pd Ag/C≥PdCo/C>PdFe/C>PdCu/C>Pd/C。其中,最优的PdZn/C在0.35~0.5 V(vs. Ag/AgCl)电压下的ORR比活性比Pt/C高3倍。除了金属元素外,引入非金属元素同样可以提高合金的催化性能[295–299]。Sampath等[296]制备了少层MoS2(1–x)Se2x合金,其活性高于原始MoS2和MoSe2。通过调节MoS2(1–x)Se2x中Se/S的掺入比例,系统地研究了催化剂的构效关系,并发现MoS1.0Se1.0显示了最高的HER活性。Gong和Xu课题组通过对Mo-S-Se合金的研究,发现了类似结果[297,299]。He等[298]成功地控制了碳纤维上三元WS2(1–x)Se2x纳米管中硫和硒的组成,所造成的无序原子排列使WS2(1-x)Se2x具有优异的电催化性能。Jin等[300]进一步获得了三元黄铁矿型CoPS用于光/电化学析氢反应。由于CoPS中P2–配体具有更高的给电子性质,该三元黄铁矿CoPS具有更适中的氢吸附能力,使得其活性高于CoS2。
4. 结论与展望
电化学氢-水转化过程中的大部分能量耗散是由能源系统中的驱动电化学反应的活化能引起的。由于催化材料对反应活化能的重要影响,催化材料是高效能源系统的核心。为了实现一个高效、可持续的能源系统,迫切需要加快开发低成本、高活性的电催化剂。一般来说,催化剂的性能取决于两个主要因素:①给定区域内活性位点的数目;②每个活性位点的本征活性。调整几何结构(即纳米构筑和晶面工程)可有效地增加活性位点的密度。当催化剂尺寸减小到纳米尺度时,催化剂具有高的比表面积,并且增加催化活性晶面的暴露。缺陷工程和非晶化也可以通过暴露热力学不稳定活性位或不饱和原子位点来增加活性位点的数量。提高活性位点的本征活性是提高活性的一个更基本但更困难的策略,因为这涉及对催化材料电子结构的精确调节。本征活性的优化需要对反应的特性有基本理解和对具有目标功能的催化剂的设计有深刻的见解。本文所讨论的典型实例(其催化性能总结见表2至表4)应用理论与实验研究相结合,通过元素掺杂、界面工程、晶相工程、合金化等手段,成功地调节了催化剂的本征活性。
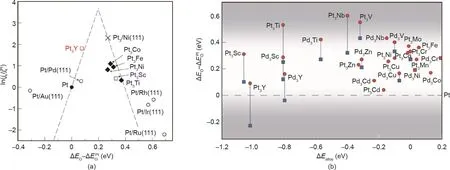
图20. (a)测试所得动力学电流密度和计算所得氧吸附能ΔEO之间的关系;(b)铂基合金表面铂的ΔEO对合金能量图[293]。
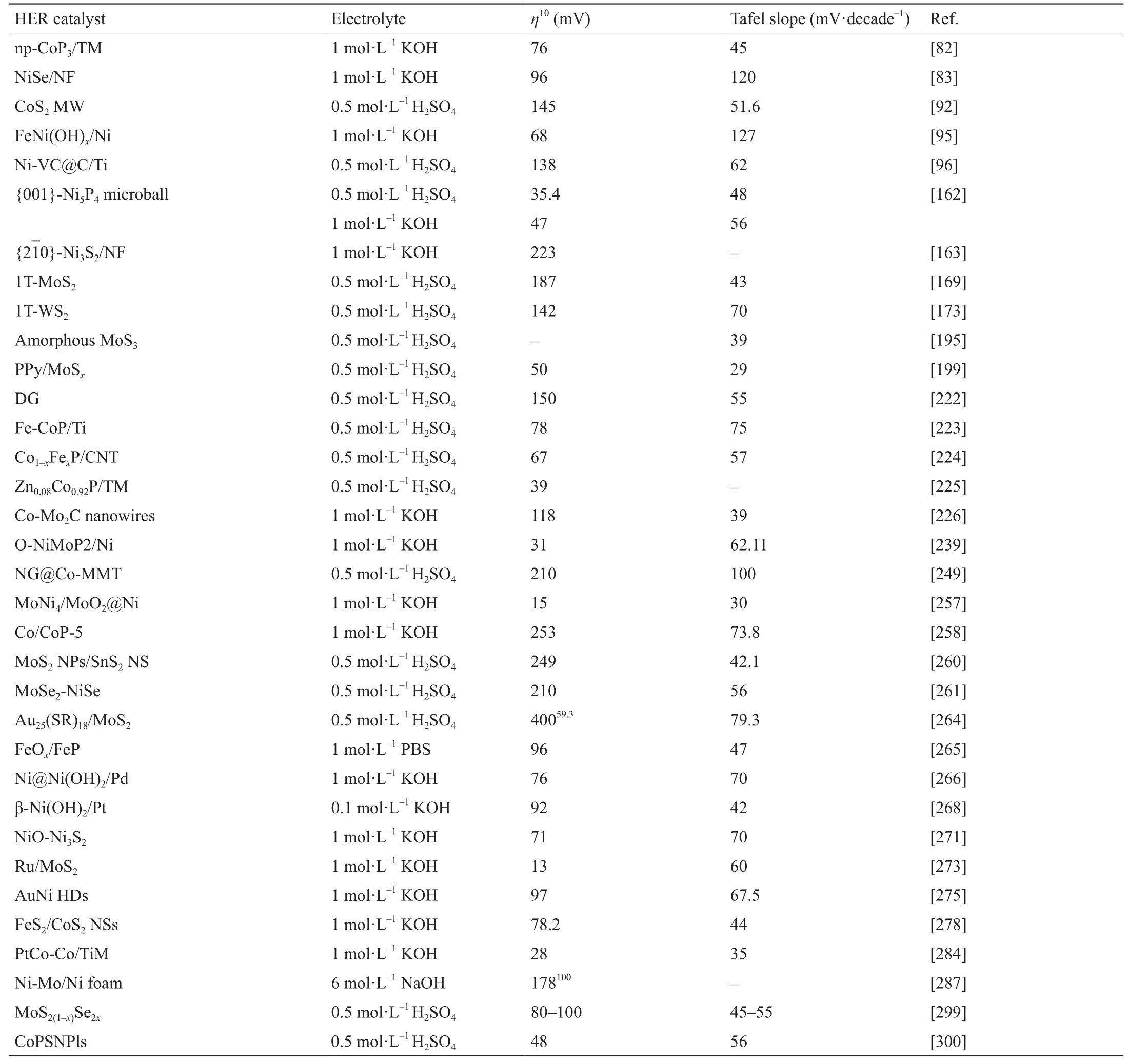
表2 典型HER催化剂性能
目前,随着化学合成技术的飞速发展,许多催化材料以其优异的电催化性能被报道。同时,物理表征方法和理论计算的发展也为研究人员提供了更多的依据和指导,使我们更好地了解催化剂的性能提升的机制。然而,现在电极催化剂的生产仍主要是通过传统的试错工艺,而不是通过合理的设计。这种研究模式不仅浪费了社会资源和科研人员的精力,而且极大地延缓了科学发展。因此,迫切需要从最基本的层面发展一种面向性能的电催化剂设计策略,并使之能应用于实际的催化剂设计合成中。这个目标需要计算化学、电催化化学和合成化学协同工作;然而不幸的是,目前这三个领域都需要改进。目前的理论计算模型还不完善,因为大多没有考虑离子强度、双层效应、溶剂化效应等因素的影响,难以准确反映催化材料表面的实际反应过程。此外,尽管这些氢和氧参与反应的机制已经被广泛研究,但在不同催化剂表面上的实际反应机制仍然是个谜。事实上,由于大多数催化剂的表面结构在电催化过程中一直在变化,因此很难确定实际的活性中心。先进的理论计算和实验表征(如原位、非原位和operando技术)的结合将带来新的进展,这将有助于我们在分子水平上理解电化学反应机制和催化剂的动态演化。最后,面向功能的催化剂制备仍是一个重大挑战。现有的材料合成技术能够在纳米尺度上对特定催化剂的理化性质进行一定程度的调控,但利用可控合成技术在原子尺度上调控催化剂的电子结构还不成熟。发展适用性强、规模化生产的可控合成方法是未来研究的另一个热点。
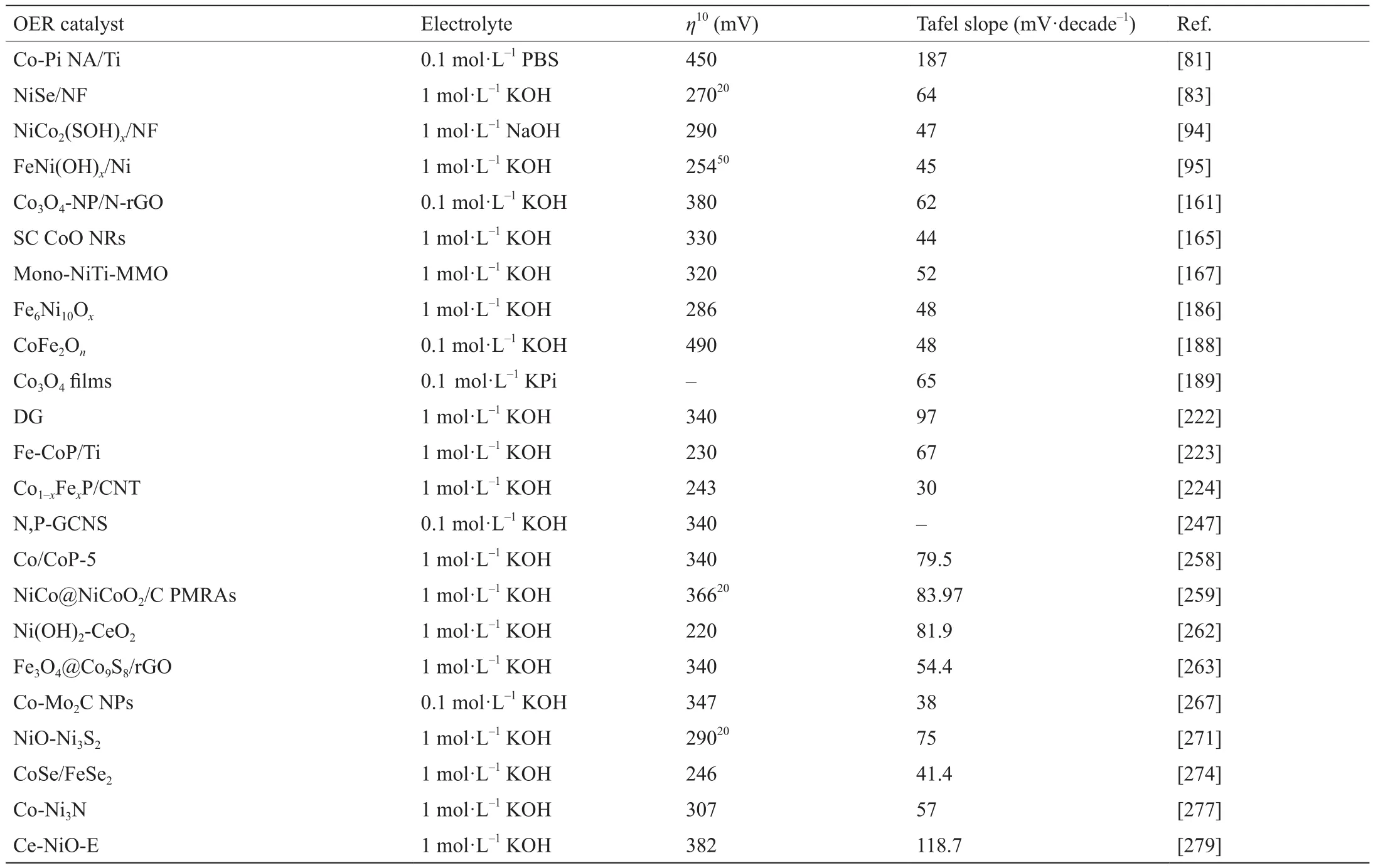
表3 典型OER催化剂性能

表4 典型非贵金属ORR催化剂性能
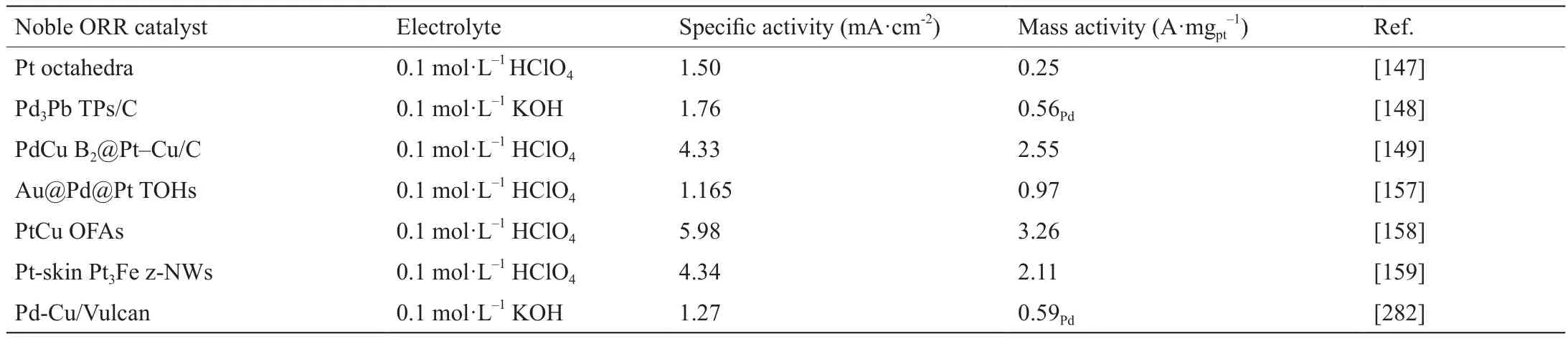
表5 典型贵金属ORR催化剂性能
致谢
感谢国家自然科学基金项目(No. 21576032,51772037)、国家自然科学基金重点项目(No. 21436003)、国家自然科学基金重大研究计划(No. 91534205)和国家基础研究重点项目(No. 2016YFB0101202)的资助。
Compliance with ethics guidelines
Lishan Peng and Zidong Wei declare that they have no conflict of interest or financial conflicts to disclose.
Nomenclature
creactant surface concentration, mol·m–3
FFaraday constant, ≈96 485 C·mol–1
GGibbs free energy, J·mol–1
fdecay rate to products and reactants, dimensionless
icurrent, A
jcurrent density, A·cm–2
j0exchange current density, A·cm–2
nnumber of electrons transferred in the reaction, dimensionless
Rresistance, Ω
Ttemperature, K
U0reaction equilibrium potential, V
Vvoltage, V
acharge transfer coefficient, dimensionless
lovervoltage, V
ΔGactactivation energy barrier, J·mol–1
ΔGH*hydrogen adsorption free energy
ΔGmaxgibbs free-energy change, J·mol–1
ΔGO*oxygen adsorption strength
