磁化TEVA树脂的制备及其表征
2020-08-21陶苗苗康海英常志远郑维明谈树苹
张 彤,陶苗苗,康海英,常志远,郑维明,谈树苹
中国原子能科学研究院 放射化学研究所,北京 102413
在PUREX流程中,为控制锝的走向,需对关键工艺点的锝浓度进行分析[1]。目前国内分析高放样品中锝含量的方法为热室内稀释-手套箱内液液萃取分离-液闪测量[2]。该方法操作繁琐耗时较长,人员受照剂量较大,且液闪测量产生的废液难于处理。
X射线荧光光谱(XRF)法可测量固体、液体及粉体,测量快速、精密度高,操作步骤简单,且设备简单,易于密封,在后处理分析中具有显著优势。但相比于其它测量方法,XRF检出限高,在后处理样品分析中存在一定局限性。
使用磁助制样(MASP)方法可有效降低XRF方法的检出限,该方法使用顺磁性分离材料,将样品中的待测组分选择性吸附至固相,通过磁场进行固液分离,完成制样。使用XRF直接分析磁助分离后的顺磁性分离材料,从而计算出样品中待测组分的浓度。相比于其他富集制样方法,磁助制样操作简单、快速,产生二次废物较少,可有效降低方法的检出限。
磁助制样方法的核心是制备顺磁性分离材料,目前制备顺磁性分离材料的常用方法主要有4种:核-壳复合磁颗粒制备法[3-4]、磁性萃取色层树脂制备法、磁性萃淋树脂制备法[5]以及化学转化磁化树脂法。目前研究较多的为核-壳复合磁颗粒制备法。该方法使用萃取剂包裹磁性颗粒或在氧化硅等载体上包裹磁颗粒制备复合颗粒[6-7],为获得高比表面积,颗粒粒径一般为纳米级。但用于XRF制样时纳米材料粒径太小,操作难度较大。化学转化磁化树脂方法对树脂的化学分离性能影响较小,可在保留树脂的选择性吸附性能的同时为树脂附加磁性,达到快速磁分离的效果,方法操作简单,适用面广,设备要求低,反应时间短。吴雪辉等[8-9]使用磁化树脂方法制备了磁性大孔交换树脂,制得磁化树脂与原树脂性能基本一致,但耐酸性较差,而后处理样品普遍为酸性介质,故难以在后处理分析中推广应用。推测其耐酸性较差原因可能是使用大孔树脂,酸易与孔道中的铁氧化物反应所致。
因此为建立磁助制样/XRF分析后处理样品中微量锝的方法,本工作拟制备对锝有选择性吸附、具有一定耐酸性能的顺磁性分离材料。综合已有的制备方法,本工作拟使用共沉淀法磁化TEVA树脂制备顺磁性分离材料用于磁助制样/XRF分析后处理中的微量锝。
1 实验部分
1.1 材料选择
1.2 试剂和仪器
所用试剂均为市售分析纯。TE-B200-S型TEVA树脂,Triskem公司。高锝酸根溶液,中国原子能科学研究院提供,c(HNO3)=0.1 mol/L,经液闪标定后Tc元素质量浓度为25.5 mg/L。
Tri-Card 2910TR型液闪谱仪,美国Perkin Elmer公司;S2 Ranger型能量色散X荧光分析仪、D8 Advance型X射线衍射(XRD)谱仪,德国Bruker公司;JSM-6360LV型扫描电镜,日本电子公司;Nicolet-iS50型红外谱仪,美国Thermo Scientific公司;Star System型TGA/DSC谱仪,瑞士Mettler Toledo公司。
1.3 磁化TEVA树脂的制备方法
将TEVA树脂浸泡在一定浓度的Fe2+和Fe3+的混合盐溶液中,滴加NaOH溶液至溶液pH值达到11,待反应完成后将磁化树脂从溶液中分离,反复冲洗晾干后得到磁化TEVA树脂。
2 结果与讨论
2.1 影响制备的主要因素
2.1.1Fe2+和Fe3+的浓度比 改变Fe2+和Fe3+的比例主要影响反应进程,对树脂的化学分离性能影响较小,对磁化树脂的磁性有一定影响。c(Fe2+)/c(Fe3+)=1~5时,在溶液中逐滴加入NaOH生成沉淀时,首先生成红棕色沉淀,继续滴加,溶液变为黑色。在18 ℃时,Fe(OH)3的溶度积为1.1×10-36,而Fe(OH)2的溶度积为1.64×10-14 [11],二者相差22个数量级。因此滴加碱溶液首先生成Fe(OH)3的红棕色沉淀,继续滴加NaOH时生成Fe(OH)2,生成的Fe(OH)2和Fe(OH)3反应生成Fe3O4,溶液颜色变为黑色。在c(Fe2+)/c(Fe3+)=10时,在溶液中滴加NaOH,首先生成Fe(OH)3红棕色沉淀,继续滴加NaOH,溶液变为墨绿色,继续滴加至pH值达到11,陈化后溶液变为黑色。继续滴加NaOH时生成Fe(OH)2过量,未反应完的Fe(OH)2为绿色,与溶液中Fe3O4混合,故溶液呈墨绿色。陈化过程中过量Fe(OH)2被氧化为Fe3O4,溶液颜色变为黑色。在c(Fe2+)/c(Fe3+)>30时,在溶液中滴加NaOH,溶液变为墨绿色,此时溶液中含有大量Fe(OH)2,继续滴加至pH值达到11,陈化后溶液变为黑色。部分Fe2+和Fe3+的浓度比对磁化TEVA树脂的影响数据列于表1。对比表1,由于总铁盐浓度不变而Fe2+浓度远高于Fe3+浓度,因此Fe2+浓度基本一致而Fe3+浓度不同。当加入少量Fe3+后,磁化TEVA树脂中的铁氧化物含量显著增加,磁助分相时间缩短,说明磁性增强。分析其原因,Fe(OH)3易于和其他粒子结合或发生离子交换形成共沉淀,是应用较广的沉淀剂、载带剂[12]。通过滴加NaOH,生成的Fe(OH)3与溶液中TEVA树脂结合,继续滴加NaOH生成的Fe(OH)2与树脂上的Fe(OH)3反应生成Fe3O4,此时Fe3O4镶嵌在树脂表面,增加了磁化TEVA树脂的磁性。若溶液中缺少作为桥梁的Fe(OH)3,则减少了树脂中Fe3O4的合成。因此,磁化过程中,加入Fe3+生成Fe(OH)3沉淀对增加磁化TEVA树脂的铁氧化物的含量及磁化TEVA树脂的磁性有重要意义,故选择使用c(Fe2+)/c(Fe3+)=1~5的条件下制得的磁化TEVA树脂用于磁助制样。

表1 部分Fe2+和Fe3+的浓度比对磁化TEVA树脂的影响Table 1 Effects of Fe2+ and Fe3+ concentration ratio on magnetic TEVA resin
2.1.2混合铁盐浓度的影响 改变混合铁盐浓度(c(Fe))对树脂的化学分离性能影响较小,对磁化TEVA树脂的磁性有一定影响,结果列于表2,其中c(Fe2+)/c(Fe3+)=1∶1不变。由表2可知,使用铁盐浓度为0.5 moL/L条件下制备的磁化TEVA树脂磁性较弱,进行磁助制样时,分相时间为5 s,将上层清液倒出时有少量颗粒流出,制得样品较松散,对测量精度有一定影响。随着铁盐浓度提升,磁化TEVA树脂中铁氧化物含量增加,磁性增强。但铁盐浓度提高至1.0 mol/L后磁性基本不变,原因是溶液中产生的Fe(OH)2、Fe(OH)3及Fe3O4在浓度过高时易在溶液中发生自聚。而且铁盐浓度过高时溶液粘度增大,不利于磁化TEVA树脂与溶液的分离,因此制备时混合铁盐浓度应控制在1 mol/L左右。

表2 混合铁盐浓度对磁化TEVA树脂的影响Table 2 Effects of mixed iron concentration on magnetic TEVA resin
2.2 优化后的制备方法
共沉淀法制备磁化TEVA树脂方法如下:
1) 选用粒径为50~100 μm的TEVA树脂浸泡在Fe2+和Fe3+的混合盐溶液中,溶液中混合铁盐浓度为1.0 mol/L,Fe2+和Fe3+的浓度比为1∶1~5∶1,加热溶液至40 ℃;
2) 滴加NaOH溶液至溶液pH值达到11,陈化40 min;
3) 加入酸将溶液调至中性或弱酸性后使用合适孔径的筛板抽滤将磁化树脂从溶液中分离;
4) 使用稀硝酸和去离子水反复冲洗;
5) 室温晾干,得到磁化TEVA树脂。
2.3 磁化树脂表征
2.3.1扫描电镜图 TEVA树脂及磁化TEVA树脂的扫描电镜图示于图1。从图1可以看出,在1 000倍以下观察磁化TEVA树脂表面与TEVA树脂没有明显区别,并未形成包覆结构,而在5 000倍以下观察磁化TEVA树脂表面出现颗粒,TEVA树脂中则没有。图2为放大5 000倍后的SEM图,对图2中局部区域的分析结果列于表3。由表3可知,图2中表面较为亮的点为铁氧化物,大小为纳米级,分布较为分散,含量较少。由此可以看出共沉淀法生成的铁氧化物与TEVA树脂结合时,铁氧化物颗粒很小,含量较少,分布较为分散,推测其对TEVA树脂的化学分离性能影响较小。

图1 TEVA树脂(a)和磁化TEVA树脂(b)的SEM图片Fig.1 SEM images of TEVA resin(a) and magnetic TEVA resin(b)

图2 磁化TEVA树脂的SEM图片Fig.2 SEM images of magnetic TEVA resin
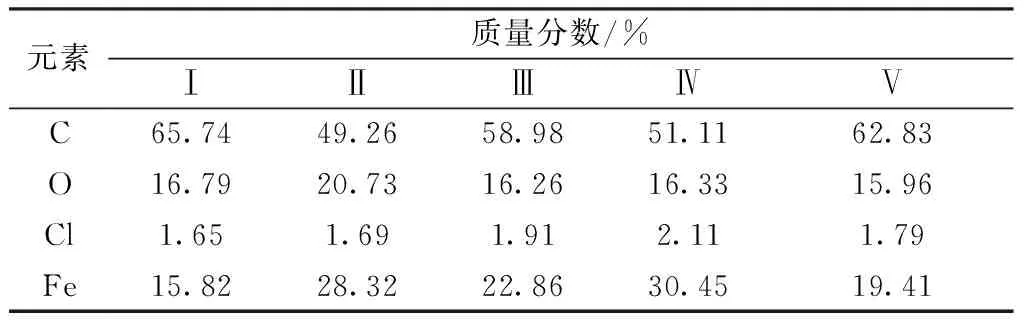
表3 磁化TEVA树脂局部碳、氧、氯和铁元素含量Table 3 Carbon, oxygen, chlorine and iron mass fraction of magnetic TEVA resin partial

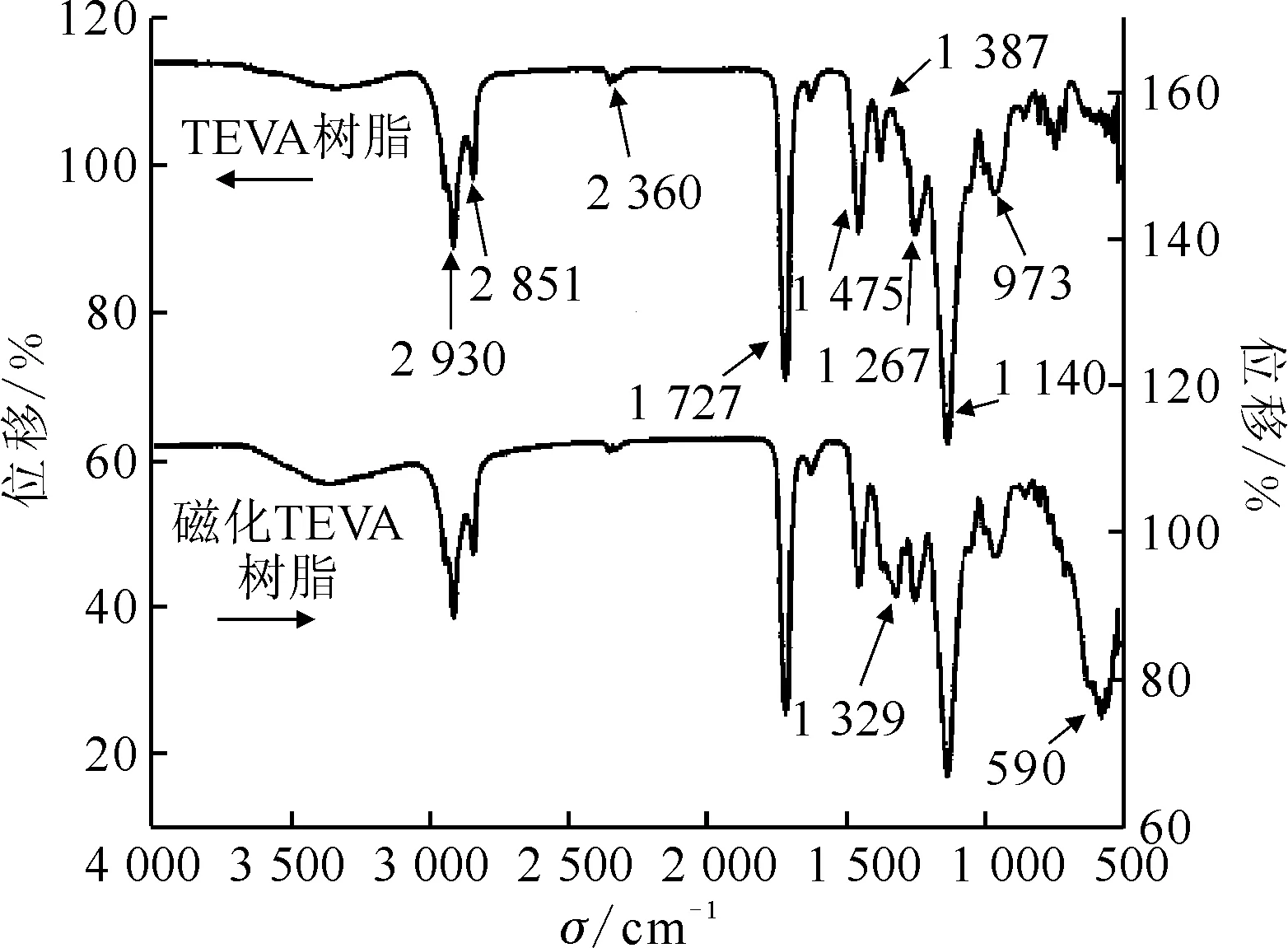
图3 TEVA树脂和磁化TEVA树脂的红外谱图Fig.3 FTIR of TEVA resin and magnetic TEVA resin
2.3.3XRD 谱图 由于磁化TEVA树脂中,铁氧化物与树脂结合较为紧密,因此将磁化TEVA树脂密堆积后直接测量。TEVA树脂及磁化TEVA树脂的电子衍射图示于图4。对比图4可知,在TEVA树脂的XRD谱图中,没有出现Fe3O4标准峰;而磁化TEVA树脂的XRD谱图中,在Fe3O4标准峰位出现了明显的尖峰。经MDI Jade软件解谱确认,磁化TEVA树脂中铁氧化物主要为Fe3O4。
2.3.4TGA/DSC 谱图 TEVA树脂和磁化TEVA树脂的热重分析和差热分析数据示于图5。对比图5可知,磁化TEVA树脂和TEVA树脂的TGA/DSC曲线基本一致。升温超过65 ℃时吸收少量热量,可能发生分解。温度超过200 ℃后开始热解碳化,放出大量热量,温度超过300 ℃后剩余碳开始被氧气氧化,继续放热,超过500 ℃后趋于一定值。TEVA树脂及磁化TEVA树脂中,有机萃取剂及骨架随着温度的升高会逐步被氧化分解,而磁化TEVA树脂中的铁氧化物不会,二者谱图一致说明磁化过程中加入的铁氧化物和TEVA树脂在加热过程中没有较为明显的吸热放热反应,印证了磁化生成的铁氧化物是物理镶嵌在TEVA树脂表面,没有发生化学反应。由TGA曲线计算磁化TEVA树脂中的铁氧化物质量分数约为7%。
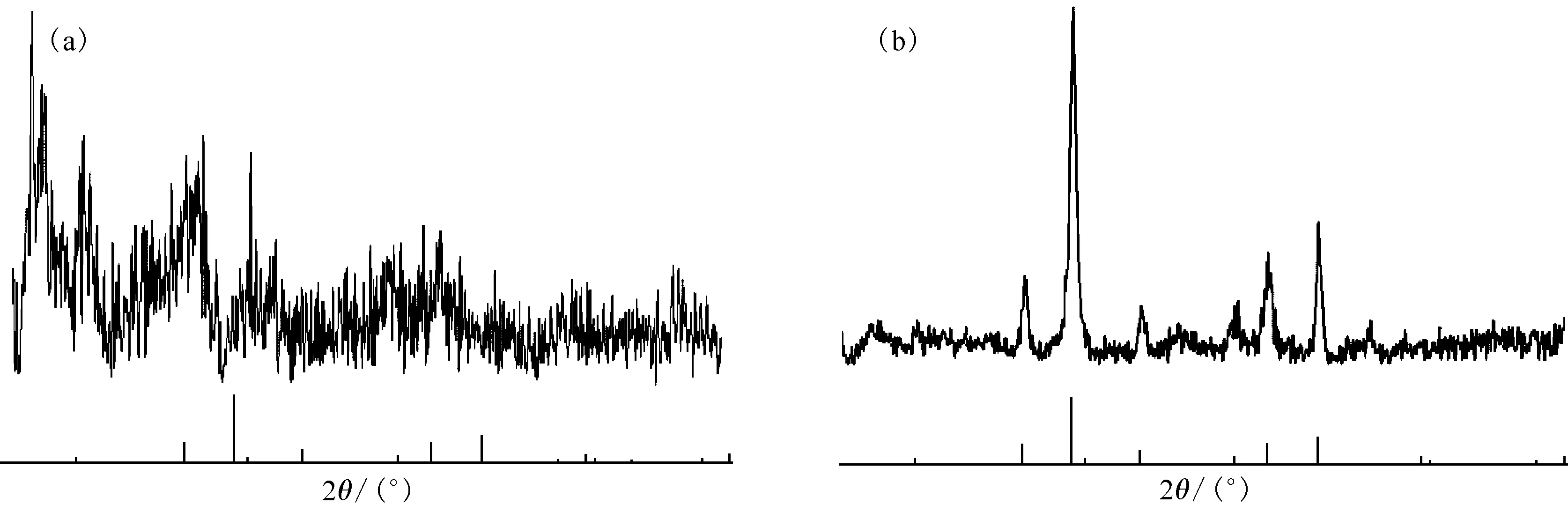
谱图中下面部分竖线为Fe3O4标准出峰位置图4 TEVA树脂(a)和磁化TEVA树脂(b)的XRD图Fig.4 XRD patterns of TEVA resin(a) and magnetic TEVA resin(b)
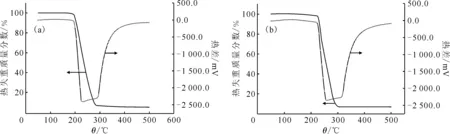
升温速率为100 ℃/min图5 TEVA树脂(a)及磁化TEVA树脂(b)的TGA/DSC谱图Fig.5 TGA/DSC curves of TEVA resin(a) and magnetic TEVA resin(b)
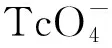
Q′=Q/(1-w(FeO))
式中:Q为磁化TEVA树脂的饱和吸附容量;Q′为修正磁化TEVA树脂的饱和吸附容量。修正磁化TEVA树脂的饱和吸附容量即扣除磁化TEVA树脂中铁氧化物的增重对饱和吸附容量测量的影响,只对比磁化过程对树脂中有效组分的化学分离性能影响。选用TEVA树脂吸附Th的饱和吸附容量设计值为70 mg/g,实测为(64±3) mg/g(n=7)。经计算,磁化TEVA树脂的饱和吸附容量为54 mg/g,铁氧化物质量分数为7%,修正饱和吸附容量为58 mg/g,磁化后树脂的饱和吸附容量下降约10%,磁化过程对树脂的饱和吸附容量影响较小。
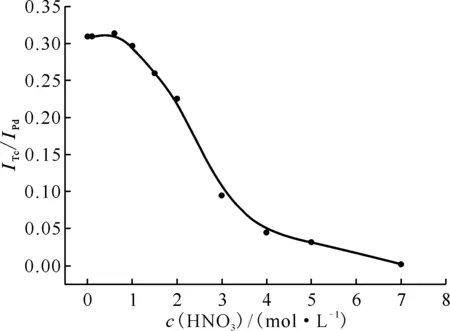
ITc、IPd分别为待测元素Tc和靶元素Pd的特征信号,以Pd作为内标,ITc/IPd作为Tc信号的观测值0.1 g磁化TEVA树脂,锝质量浓度为2.55 mg/L的5 mL稀硝酸溶液,反应时间5 min图6 HNO3浓度对Tc测量的影响Fig.6 Effect of HNO3 concentration on Tc analysis
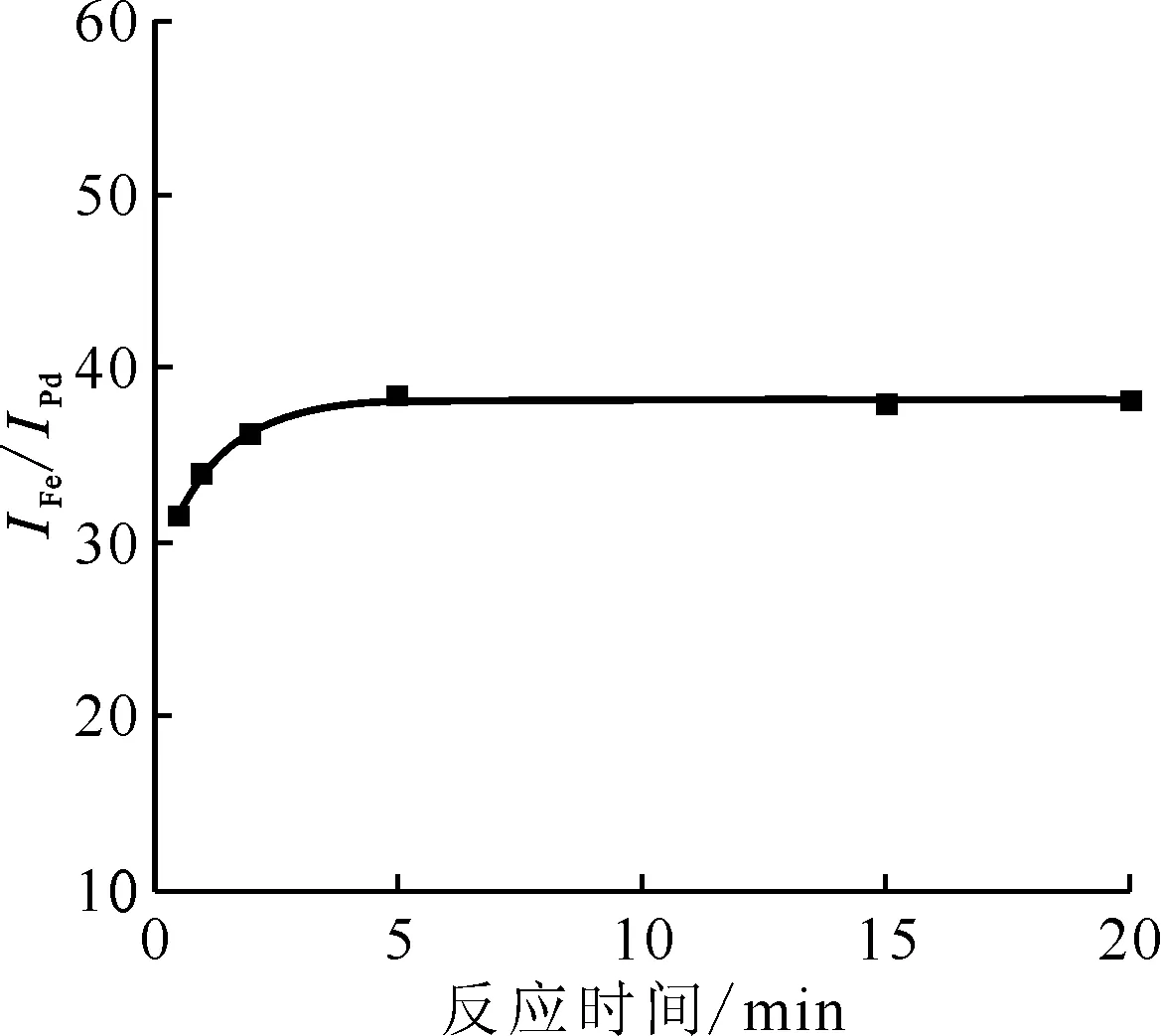
IFe、IPd分别为待测元素Fe和靶元素Pd的特征信号,以Pd作为内标,IFe/IPd作为Fe信号的观测值0.1 g磁化TEVA树脂,5 mL 0.1 mol/L硝酸溶液图7 反应时间对Fe信号的影响Fig.7 Effect of reaction time on iron signal
2.4 测试
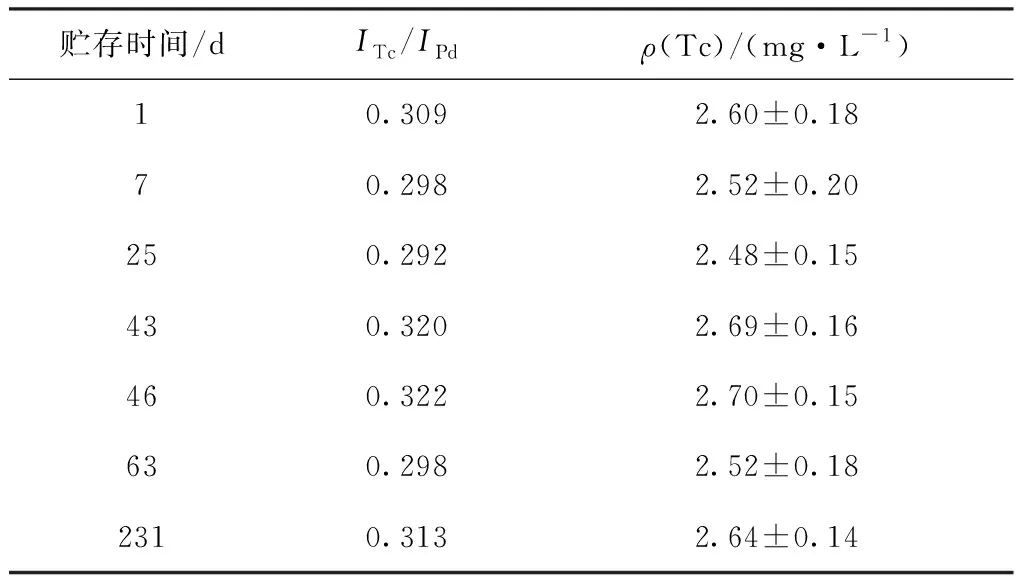
表4 磁化TEVA树脂的稳定性Table 4 Magnetic TEVA resin stability
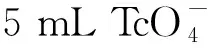

表5 制备方法的重复性Table 5 Repeatability of preparation methods
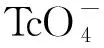
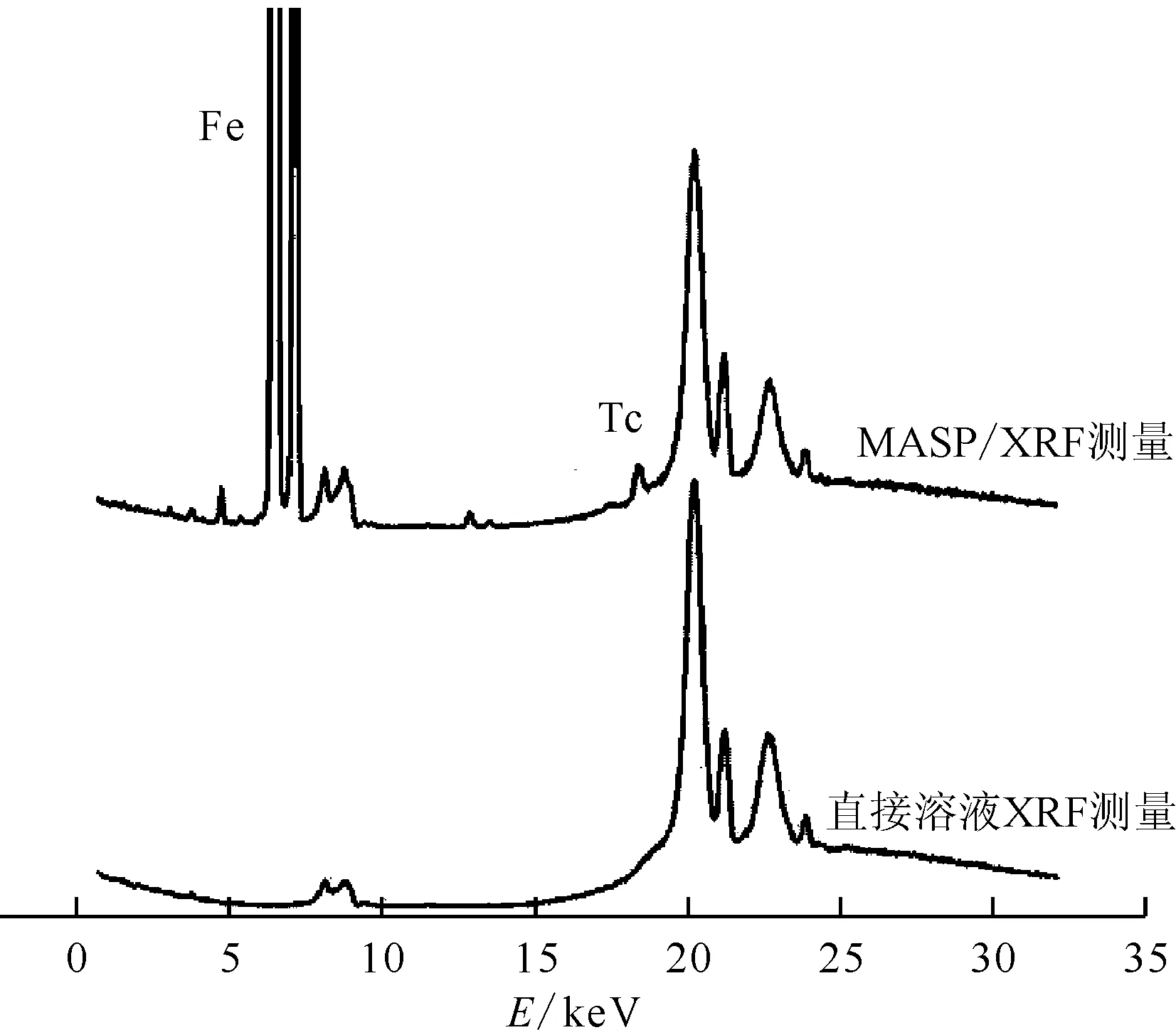
5 mL 0.1 mol/L硝酸溶液质量浓度为2.55 mg/L)图8 XRF磁助制样测量和直接溶液测量谱图Fig.8 XRF spectrometry of MASP and aqueous sample
3 结 论
使用共沉淀方法磁化TEVA树脂,通过扫描电镜、红外、XRD等方法进行表征。结果显示磁化TEVA树脂既保留了TEVA较好的化学分离性能,又具备了顺磁性便于快速分离。该制备方法操作简单、重复性好,制得的磁化TEVA树脂性能稳定,具有一定耐酸性能,可用于磁助制样/XRF分析后处理样品中的微量锝。
使用该树脂所建立的磁助制样/XRF分析方法可有效降低方法检出限,并降低后处理样品中复杂组分对微量锝分析的影响,有望用于实际工艺点样品的测量。
