一测多评法测定甜叶菊中6种绿原酸类成分的含量△
2020-08-21张民达刘梦婷李小莹刘秀斌曾建国
张民达,刘梦婷,李小莹,刘秀斌,曾建国
1.湖南农业大学 中兽药湖南省重点实验室,湖南 长沙 410128;2.湖南农业大学 园艺园林学院,湖南 长沙 410128;3.湖南农业大学 动物医学院,湖南 长沙 410128
甜叶菊Steviarebaudiana又名甜草、甜菊、甜茶,是一种菊科多年生草本植物。甜叶菊含有多种功能性成分,包括糖苷类、黄酮类、绿原酸类等。甜叶菊糖苷类化合物种类最多、含量最大,具有极高的甜度,其中甜菊苷甜度为蔗糖的300倍[1],作为甜味剂在食品、药品、化工、酿酒等行业中广泛使用。目前甜叶菊主要作为生产的甜菊糖苷的原料,而针对绿原酸等活性成分开发利用的研究较少。
绿原酸类多酚成分是由咖啡酸与奎尼酸生成的缩酚酸,为金银花和山银花中抗菌、抗病毒的主要有效成分。随着对绿原酸研究的不断深入,发现其具有抗氧化、抗肿瘤、抗菌、抗病毒、免疫调节、降糖等多种活性作用[2]。目前,已在甜叶菊中发现包含咖啡酰奎宁酸、二咖啡酰奎宁酸和三咖啡酰奎宁酸在内的24种绿原酸类化合物[3];另外,甜叶菊中还含有具有抗肿瘤、抗炎、抗氧化、降血糖等生物活性的槲皮苷等黄酮类成分[4]。绿原酸已经作为一种饲料添加剂被广泛应用于畜牧、水产养殖等行业中[5],而异绿原酸作为饲料添加剂在代替抗生素使用、提高饲料利用率及加强生产性能等方面具有良好前景[6]。从甜叶菊中生产甜菊糖苷的同时,开发利用绿原酸和槲皮苷等活性成分,这为实现甜叶菊资源的综合利用和低成本获得绿原酸和槲皮苷的原料来源提供了新的思路。由于多指标的质量控制模式需要对照品的种类和数量均比较大,且部分对照品价格比较昂贵且难以获得,王智民等[7]提出了一测多评的多指标质控模式,即在多指标质量控制时,以样品中对照品廉价易得的常见成分为内标,建立该成分与其他成分间的相对校正因子(Relative Correction Factor,RCF),再通过校正因子计算出其他成分的含量。在方法实施时,可在只有1个对照品而其余对照品不足的情况下,实现这些成分的同步含量测定,并已成功应用于苦参、丹参注射液、黄柏、附子、双黄连口服液等中药的质量控制[8]。其中黄连药材的一测多评法(QAMS)被收录于2010年版《中华人民共和国药典》[9]。付晓等[10]采用高效液相色谱法(HPLC)同时测定甜叶菊中3种绿原酸类化合物,为了更加全面地评价甜叶菊原料及提取物中绿原酸和槲皮苷等有效成分的含量,本实验利用HPLC以绿原酸为内标,采用斜率校正法建立其与甜叶菊中其他5种绿原酸类成分新绿原酸、隐绿原酸、异绿原酸B、异绿原酸A、异绿原酸C的相对校正因子,同时建立绿原酸与槲皮苷在相同条件下的相对校正因子。并计算各成分含量,同时采用外标法同步测定,验证QAMS得到结果的准确性和可靠性。
1 材料
Agilent 1260高效液相色谱系统、Agilent ChemStation工作站、Waters E2695、Waters ACQUITY Arc 高效液相色谱系统(USA,Waters),Empower工作站;色谱柱为Agilent ZORBAX SB-C18柱、华谱新创Unitary C18柱和Diamoncil C18柱(色谱柱规格均为250 mm×4.6 mm,5 μm);十万分之一电子天平(USA,METTLER TOLEDO)。
绿原酸(中国食品药品检定研究院,批号:110753-201415,纯度:96.20%);新绿原酸(德思特生物,批号:DST180130-015,纯度:99%);隐绿原酸(德思特生物,批号:DST180210-035,纯度:99%);异绿原酸B(德思特生物,批号:DST180130-037,纯度:99%);异绿原酸A(中国食品药品检定研究院,批号:111782-201706,纯度:97.3%);异绿原酸C(德思特生物,批号:DST180210-038,纯度:99%);槲皮苷(一飞生物,批号:F316202,纯度:98%);甲醇(色谱纯,德国 Merck公司);磷酸(色谱纯,美国 Tedia公司)。
甜叶菊样品由山东省诸城市浩天药业有限公司提供,由湖南农业大学曾建国教授鉴定为甜叶菊正品,粉碎,过40目筛。
2 方法与结果
2.1 QAMS的建立
2.1.1色谱条件 色谱柱为Agilent ZORBAX SB-C18柱(250 mm×4.6 mm,5 μm),流动相为甲醇(A)-0.1% 磷酸水(B),梯度洗脱(0~1 min,10%~25%A;1~5 min,25%~25%A;5~12 min,25%~35%A;12~13 min,35%~45%A;13~30 min,45%~45%A);体积流量1.0 mL·min-1,进样量10 μL,检测波长:0~23 min,330 nm,23~30 min,350 nm,柱温25 ℃。样品图谱和混合对照品图谱见图1。

注:A.甜叶菊样品;B.对照品;1.新绿原酸;2.绿原酸;3.隐绿原酸;4.异绿原酸B;5.异绿原酸A;6.异绿原酸C;7.槲皮苷。图1 甜叶菊样品及对照品的HPLC图
2.1.2供试品溶液的制备 取甜叶菊样品,精确称取0.5 g于50 mL容量瓶中,70%甲醇溶解,摇匀并定容,超声2 h后用70%甲醇溶液补齐液面至刻度线,过0.45 μm滤膜,即得供试品溶液。
2.1.3对照品混合对照品溶液的制备 精确称取绿原酸0.009 66 g、新绿原酸0.001 76 g、隐绿原酸0.001 60 g、异绿原酸B 0.002 11 g、异绿原酸A 0.008 13 g、异绿原酸C 0.005 36 g、槲皮苷0.002 96 g于10 mL容量瓶中,用甲醇溶解,摇匀并定容,过0.45 μm滤膜,即得对照品混合对照品溶液。
2.1.4线性系列溶液的制备 取混合对照品溶液,采用逐级稀释法分别稀释2、5、10、20、40、60、100倍,共获得7个不同浓度的混合对照品溶液。
2.1.5线性及检测限与定量限 分别精密吸取线性系列溶液上机检测,以峰面积为纵坐标,进样浓度为横坐标,绘制标准曲线。
取线性系列溶液浓度最低者,不断稀释,取1 mL置于液相小瓶中,按2.1.1方法上机检测,分别取S/N为10、3作为各个物质的定量限及检测限。见表1。
2.1.6相对校正因子 各个物质相对校正因子为其标准曲线斜率与内标绿原酸的斜率之比,计算得到新绿原酸、隐绿原酸、异绿原酸B、异绿原酸A、异绿原酸C,槲皮苷的相对校正因子分别为1.08、1.12、1.17、1.33、1.27、0.63。
2.1.7准确度试验 精密称取0.25 g样品9份于50 mL容量瓶中,按高(150%)、中(100%)、低(50%) 3个浓度各3份加入7个对照品适量,70%甲醇溶解并定容,按照2.1.2项方法提取。按2.1.1项方法进样,计算平均加样回收率。得到绿原酸,新绿原酸、隐绿原酸、异绿原酸B、异绿原酸A、异绿原酸C、槲皮苷的不同添加水平的平均回收率分别为102.74%、102.51%、103.23%、101.68%、98.11%、96.61%、99.87%。RSD分别为:6.98%、6.95%、2.93%、5.20%、6.18%、5.76%、5.09%。
2.1.8重复性试验 取样品6份,照2.1.2项下方法平行制备,进样分析。以标准曲线法计算7个化合物的含量,其RSD分别为1.05%、0.98%、1.23%、1.00%、0.99%、1.00%、1.11%。表明重复性良好。
2.1.9日内日间精密度 精密移取高、中、低3个浓度混合对照品溶液重复进样6次,测定各成分的峰面积。计算得到绿原酸、新绿原酸、隐绿原酸、异绿原酸B、异绿原酸A、异绿原酸C、槲皮苷低浓度的RSD分别为0.1%、0.1%、0.2%、0.2%、0.0%、0.1%、1.1%;中浓度的RSD分别为0.9%、1.0%、1.1%、1.2%、1.0%、1.1%、1.1%;高浓度的RSD分别为1.3%、1.3%、1.3%、1.2%、1.3%、1.2%、1.5%;精密吸收中浓度混合对照品溶液,每天进样3次,连续3 d,记录峰面积。计算得到绿原酸、新绿原酸、隐绿原酸、异绿原酸B、异绿原酸A、异绿原酸C、槲皮苷的RSD分别为2.0%、2.0%、2.3%、1.9%、2.1%、2.0%、2.1%。表明日内日间精密度良好。
2.2 耐用性
2.2.1柱温和流速耐用性 在相同条件下,分别考察不同流速(0.8、1.0、1.2 mL·min-1)和不同柱温(20、25、30 ℃)对7个物质相对保留时间的影响,结果显示不同流速和柱温对相对保留时间的影响较大(RSD>5.0%),检测过程中应当对柱温和流速严格控制以确保结果的准确。
2.2.2相对校正因子的耐用性 本实验分别选用Agilent 1260、Waters E2695和Waters ACQUITY Arc 3套高效液相色谱系统与3种不同色谱柱考察色谱系统对相对校正因子的影响。结果显示不同色谱系统和色谱柱对RCF的影响较小(RSD<4.0%,重复性良好,见表2)。
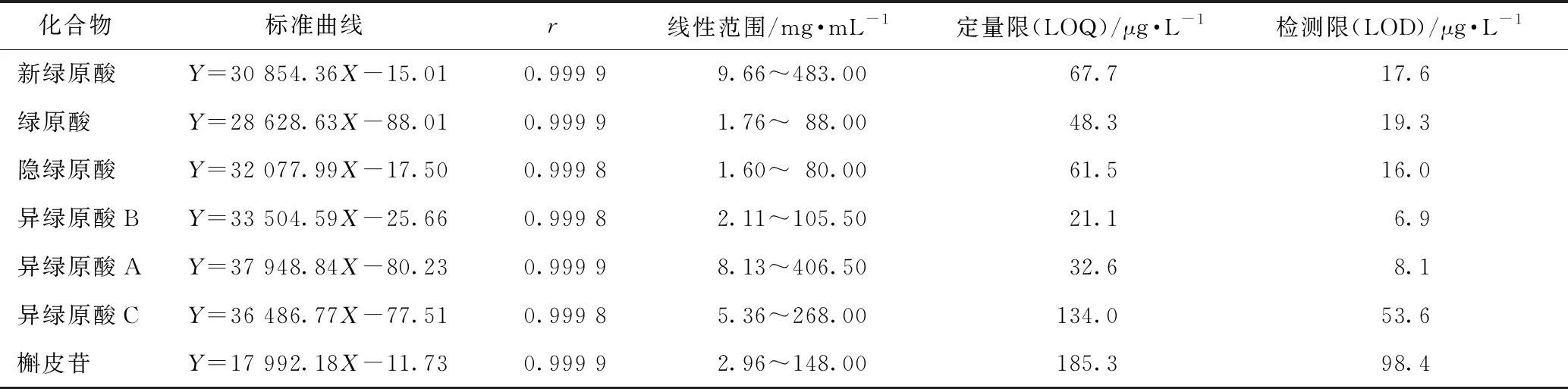
表1 6种绿原酸类成分及槲皮苷的线性及检测限与定量限
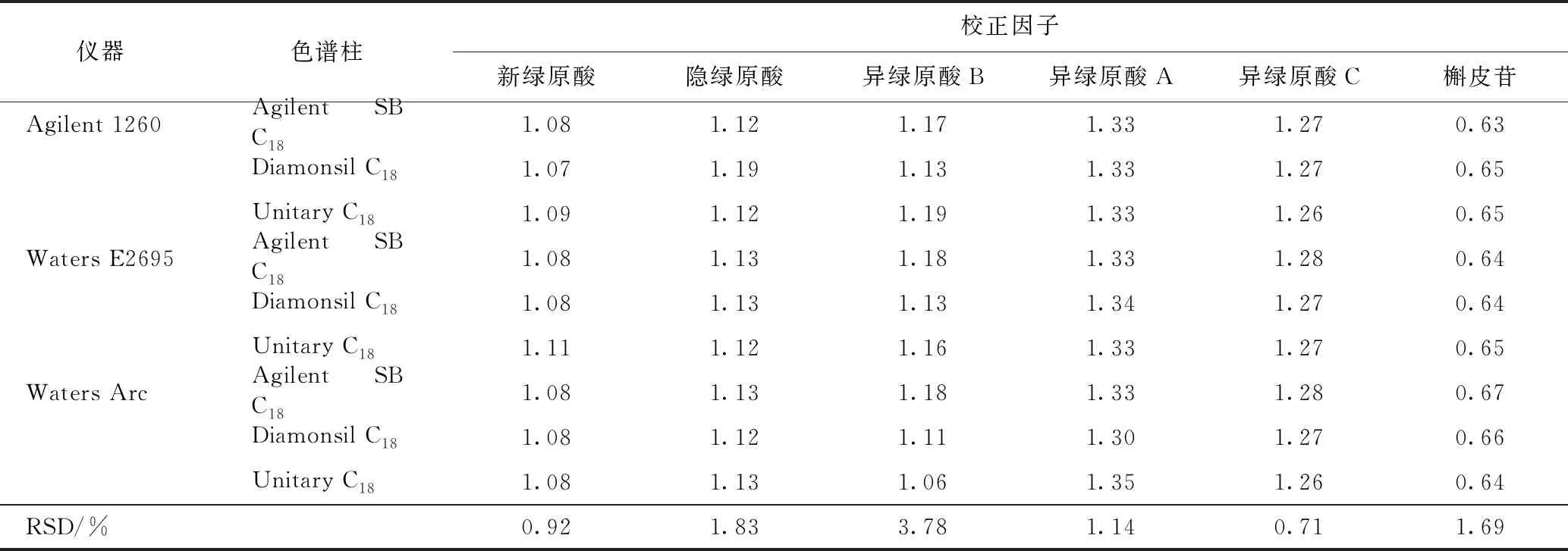
表2 一测多评法不同色谱系统与色谱柱条件下6种绿原酸成分相对校正因子的耐用性
2.3 与外标法的比较
分别采用QAMS与外标法对15批甜叶菊样品进行检测,平行测定3次。结果显示:1)外标法与QAMS结果差异有统计学意义;2)不同批次样品中7个物质的总量差异较大,各成分比例也存在较大差别。
3 讨论
3.1 建立对槲皮苷的含量测定方法
在实验过程中发现,甜叶菊中含有大量黄酮类活性成分槲皮苷。在利用绿原酸建立新绿原酸、隐绿原酸、异绿原酸B、异绿原酸A、异绿原酸C的相对校正因子的同时,也对槲皮苷的相对校正因子进行了测定,并对其进行方法学考察,结果显示此方法可以用来测定甜叶菊中槲皮苷的含量。
3.2 检测波长的选择
对绿原酸、新绿原酸、隐绿原酸、异绿原酸B、异绿原酸A、异绿原酸C、槲皮苷进行全波长扫描发现:绿原酸,新绿原酸、隐绿原酸、异绿原酸B、异绿原酸A、异绿原酸C具有相近的最大吸收波长,均在330 nm附近,故选择330 nm作为绿原酸类多酚的检测波长。而槲皮苷的最大吸收波长在350 nm处。选择350 nm作为检测波长。
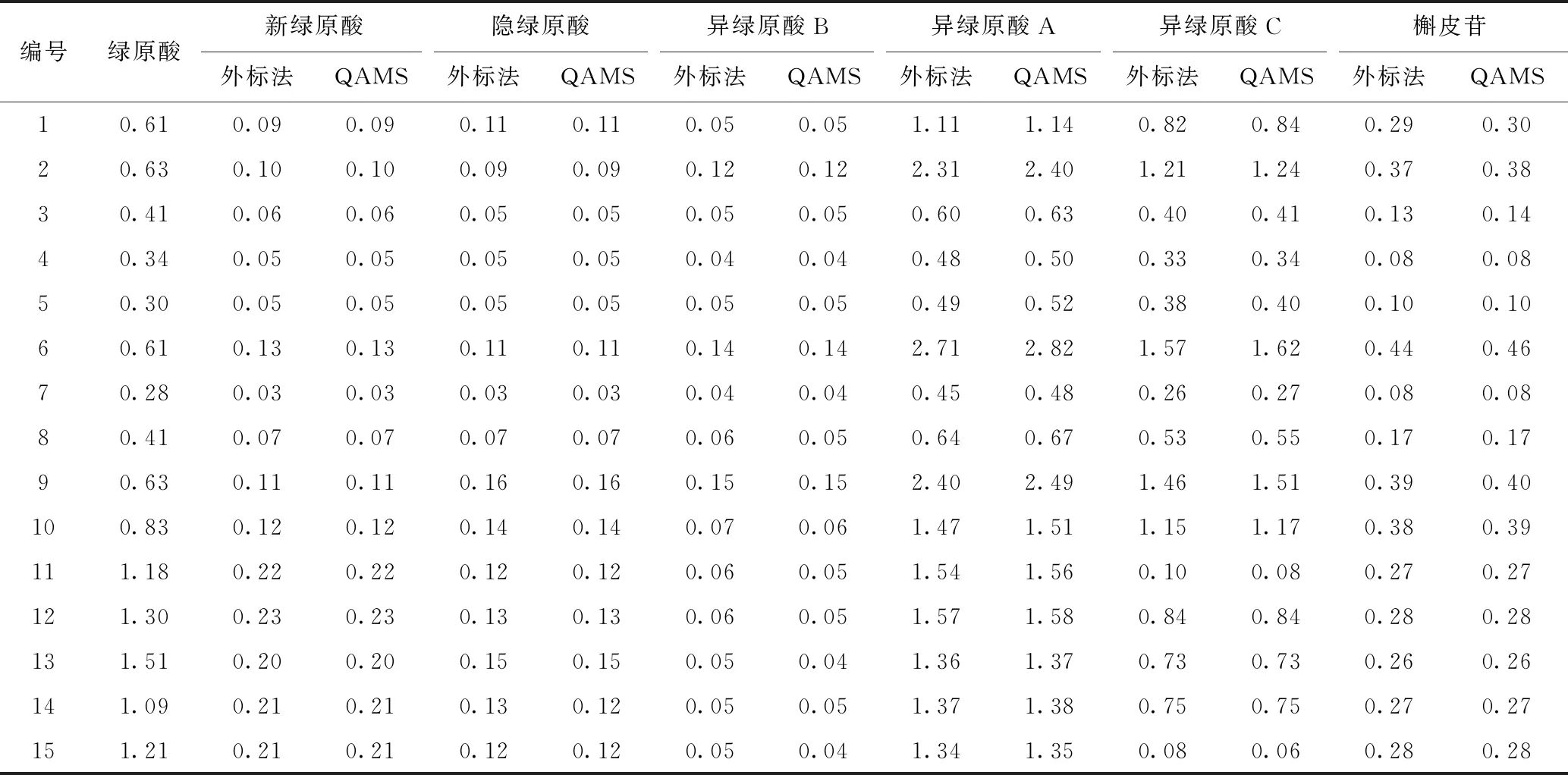
表3 15批甜叶菊样品QAMS与外标法的比较结果 %
3.3 色谱峰的定位
色谱峰的定位是通过相对保留时间来评价的。本研究考察发现柱温和流速改变对各成分相对保留时间的影响较大,不利于色谱峰的定位。但各成分的出峰顺序不受影响。在QAMS中,可以通过各成分的出峰顺序,计算待测组分与内参的保留时间差值。同时参考各成分的紫外吸收特征,基本可以确定各目标峰的位置。
3.4 质量评价
目前对甜叶菊原料的利用主要集中在甜菊糖苷的开发利用上,针对绿原酸等活性成分开发利用的研究较少。可以从甜叶菊中开发利用绿原酸和槲皮苷等活性成分,从而实现甜叶菊资源的综合利用。由于不同甜叶菊中各成分比例具有较大差异,其中任意一种成分的含量都不能正确反映甜叶菊的质量。本实验以绿原酸为内标,采用QAMS对甜叶菊叶片原料中6种成分进行含量测定,并对其进行了方法学考察,结果证明此方法在准确度、线性范围、重复性、日内日间精密度等都符合要求。考察了不同色谱仪、不同色谱柱对相对校正因子的影响,结果表明相对校正因子不受上述色谱条件的影响;同时也在此条件下对槲皮苷的含量进行了控制。采用外标法和QAMS对15批甜叶菊叶片原料进行含量测定,结果无显著性差异,证明所建立的相对校正因子定量分析方法可行,故可采用以绿原酸对照品为内标的QAMS测定甜叶菊叶片原料中绿原酸类6种活性成分的含量以评价甜叶菊原料的质量。
