CoB/CeO2催化剂的优化制备及在NaBH4液相制氢中的应用
2020-08-17刘力魁刘威李佳李芳李其明
刘力魁,刘威,李佳,李芳,李其明
(辽宁石油化工大学石油化工学部,辽宁抚顺113001)
氢气作为一种绿色环保的新型替代能源日益受到高度重视[1-6]。氢能经济发展的诸多技术环节中都不可避免地涉及氢气的高效储存,因此发展高效储氢技术成为制约氢能经济发展的关键[7-10]。
相对于传统高压物理储氢技术,化学储氢技术更具成本与安全优势[11-12]。例如,化学储氢材料NaBH4不但具有较高的储氢密度,并且可安全保存于碱性溶液中,需要氢气时通过催化剂就可以即时释放氢气,而发展高效NaBH4水解制氢催化剂成为硼氢化物液相储氢技术的关键[13-14]。过去几十年,NaBH4释氢催化剂主要集中于贵金属领域,例如,Kojima 等[15]发展了Pt/LiCoO2催化剂。Amendola 等[16]把Ru负载于离子交换树脂上制备了负载型Ru贵金属催化剂,该负载型催化剂展现了较高的催化活性。贵金属催化剂虽然催化活性较高,但是成本高,不利于推广[17],因此人们又逐步开发了诸如CoB、NiB、FeB 等过渡金属负载型催化剂[18-20],其中Guo 等[21]制备了泡沫镍负载的CoB 催化剂。Li等[22]基于开口碳纳米管催化剂载体制备了碳纳米管内腔限域的CoB 催化剂。Liang 等[23]通过磁性尖晶石NiFe2O4负载NiB 过渡金属活性组分,其目的在于提高过渡金属活性组分与催化剂载体之间的相互作用,抑制NiB脱落。研究表明,目前硼氢化物水解制氢过渡金属负载型催化剂大部分通过化学还原法制备,其是将常温化学还原的Fe、Co 和Ni 活性组分直接负载于催化剂载体,一般没有高温焙烧处理,因此过渡金属活性组分与催化剂载体的相互作用较弱,过渡金属活性组分极易脱离催化剂载体表面[24]。例如,Yu等[25]制备了TiO2、Al2O3和CeO2这3种载体负载的CoB催化剂,对比分析发现其催化活性 顺 序 为CoB/TiO2>CoB/Al2O3>CoB/CeO2>CoB,说明负载后的CoB催化剂具有更高的催化活性,然而该研究并没有涉及催化剂后处理过程对活性组分的影响。
为了提高化学还原的Fe、Co 和Ni 活性组分与催化剂载体之间的固载强度,本研究以氧化铈为模型载体,制备了一种CoB/CeO2负载型催化剂,重点探讨了处理气氛和处理温度对CoB/CeO2负载型催化剂微观结构和催化活性的影响,通过催化剂样品的对比分析阐述了CoB/CeO2负载型催化剂在不同气氛和不同焙烧温度下展示的微观形貌、晶相结构、活性组分价态和催化活性。
1 实验部分
1.1 试剂
氯化钴,天津凯通化学试剂有限公司,分析纯;硝酸铈,天津凯通化学试剂有限公司,分析纯;硼氢化钠,上海思域化工科技有限公司,分析纯;氢氧化钠,沈阳市新化试剂厂,分析纯;柠檬酸,天津市光复科技发展有限公司,分析纯;去离子水,自制。
1.2 催化剂的制备
CeO2催化剂载体的制备:称取10g Ce(NO3)3·6H2O 于洁净的烧杯中,配制成铈源溶液;分别称取6.73g 乙二胺四乙酸(EDTA)和7.26g 柠檬酸依次加入到铈源溶液中,接着向混合体系中滴加NH3·H2O 并不断搅拌,使烧杯中的物质充分混合、溶解,直至溶液的pH 接近8;将烧杯中的溶液转移至坩埚中并在电炉上加热蒸发溶液水分,随着溶液水分的不断减少,溶液趋于黏稠,形成透明黄色溶胶,溶胶持续脱水干燥,最后形成淡黄色粉末,冷却后将得到的淡黄色粉末放到马弗炉中850℃焙烧300min,得到CeO2粉体。
CoB/CeO2负载型催化剂的制备:称取0.34g CoCl2·6H2O 于洁净烧杯中,量取50mL 去离子水使其完全溶解,配制成紫红色钴源溶液,称取1.06g CeO2粉末加入到钴源溶液中,通过磁力搅拌使CeO2粉末充分混合到钴源溶液中;接着称取0.16g NaBH4溶于去离子水,得到澄清NaBH4溶液;将配制好的NaBH4溶液通过注射泵滴加至上述的CeO2-钴源混合溶液中,并持续搅拌,滴加完毕待后再静置30min。最后经过滤、洗涤、干燥,获得灰色粉末,即为新鲜制备的CoB/CeO2负载型催化剂。
CoB/CeO2负载型催化剂高温处理:将新鲜制备的CoB/CeO2负载型催化剂分别在空气、氮气中进行焙烧,设置焙烧温度分别为300℃、400℃和500℃,焙烧时间2h,并将不同载气下焙烧活化的催化剂标记 为CoB/CeO2-air(300℃)、 CoB/CeO2-N2(300℃);CoB/CeO2-air(400℃)、 CoB/CeO2-N2(400℃); CoB/CeO2-air(500℃)、CoB/CeO2-N2(500℃)。
1.3 催化剂的表征
XRD 表征采用日本理学公司D/max RB 型X 射线衍射仪(XRD),主要是对不同条件下制得的CoB/CeO2负载型催化剂进行物相分析,其实验条件为:Cu Kα辐射(λ=0.15418nm),管电压40kV,管电流30mA,使用连续扫描方式,步长0.02°,扫描速率6°/min。采用Quanta 200 FEG扫描电镜(SEM)对不同条件下制得的CeO2和CoB/CeO2负载型的微观形貌分别进行了观察。采用Perkin-Elmer PHI 5300 ESCA X 射线光电子能谱仪(XPS)进行元素分析。
1.4 NaBH4水解制氢测试
硼氢化钠水解制氢通过排水法结合Weight Lab质量测试分析软件进行测试:在加入反应物之前,将水浴温度调至反应所需温度并待其稳定,依次向反应器中加入去离子水、氢氧化钠、硼氢化钠,搅拌,使其溶解完全,最后加入适量催化剂并迅速将塞子盖好,反应开始,分析天平上即时质量会通过Weight Lab分析软件自动记录。
2 结果与讨论
2.1 催化剂的晶相结构分析
图1 分别给出了CoB、CoB/CeO2(新鲜制备)、CoB/CeO2-air (500℃)和CoB/CeO2-N2(500℃) 的XRD图。
由图1(a)可见,直接制备的CoB 材料晶相结构中没有特征衍射峰,说明直接化学还原法制备的CoB属于非晶态结构。新鲜制备的CoB/CeO2负载型催化剂,CoB/CeO2-air(500℃)和CoB/CeO2-N2(500℃)相应的衍射峰几乎完全相同,在33°、38°、55.8°和66.6°的特征衍射峰分别归属于载体CeO2萤石相(111)、(200)、(220)、(311)晶面的特征衍射[26]。对于新鲜制备的CoB/CeO2,由于常温化学还原的CoB属于非晶结构,因而其仅呈现CeO2载体的特征衍射峰。CoB/CeO2-air(500℃)和CoB/CeO2-N2(500℃)中都没有出现萤石相以外的特征衍射峰,可能有两个原因:一是CoB在相应气氛高温处理后仍然保持了非晶结构;二是CoB结构在高温处理后发生了晶相变化,然而由于含量较少,因而很难从XRD 谱图中观察到。图1(b)给出了纯CoB在氮气焙烧气氛下300℃、400℃和500℃的XRD 图,从图中可以看出纯CoB在300℃为典型非晶态结构,而在400℃出现了少量晶化衍射峰,在500℃已经很难看到非晶衍射峰,这说明CoB在高温处理时仍然出现了高温晶化。为了进一步深入分析CoB/CeO2在高温气氛处理中CoB的微观结构变化情况,对其进行了SEM和XPS分析。
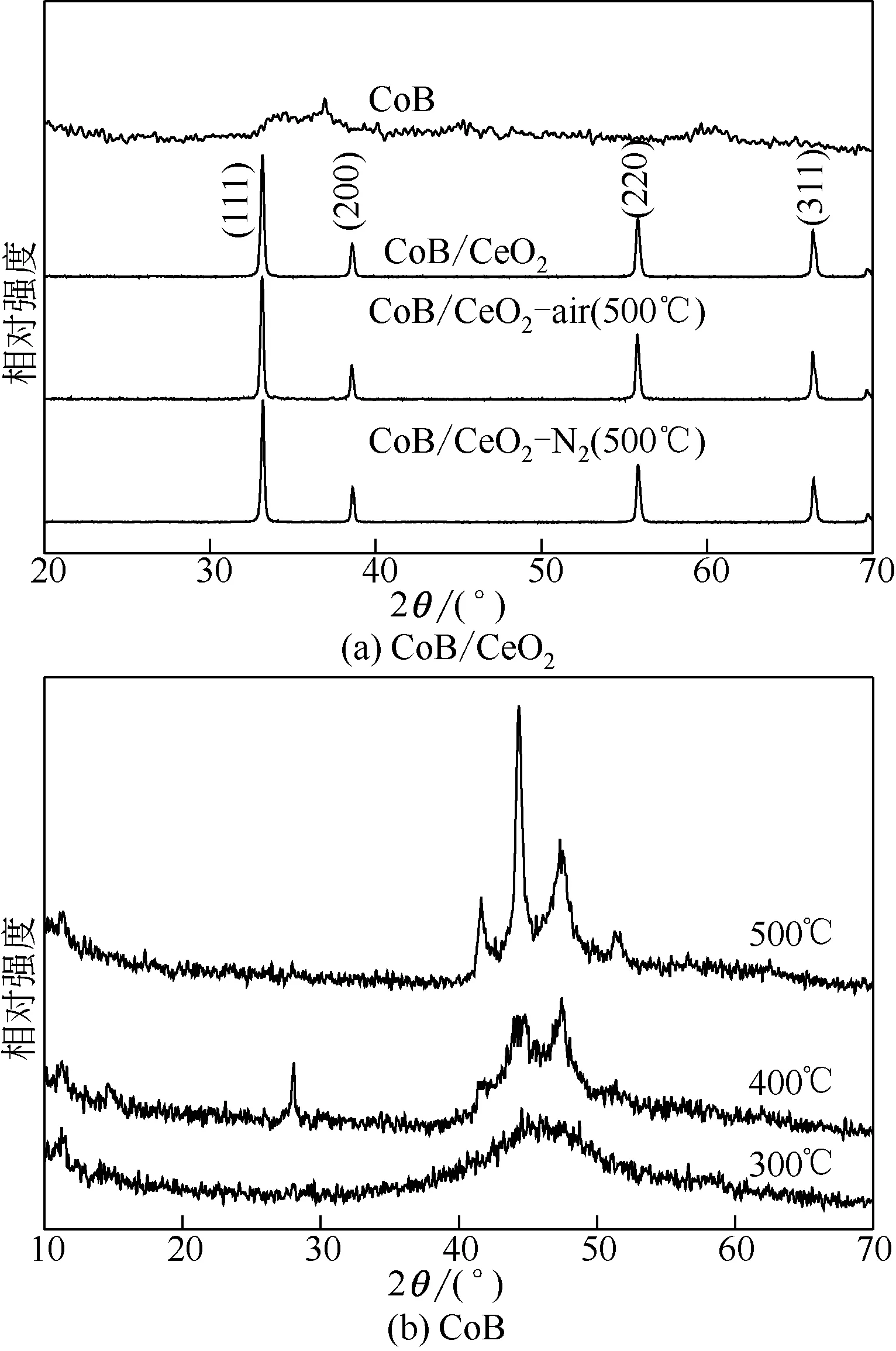
图1 CoB/CeO2和CoB催化剂在不同处理条件下的XRD图
2.2 催化剂的SEM和XPS元素分析
图2 为CeO2、CoB/CeO2、CoB/CeO2-air(500℃)和CoB/CeO2-N2(500℃)的SEM图。
图2(a)是本实验制得的CeO2载体的微观结构图,CeO2载体是由球状或类球状纳米粒子聚集而成的,形成了表面疏松多孔的蜂窝状结构,插图为纯CoB活性组分的SEM图,可以看出纯CoB磁性粒子会发生高度团聚。图2(b)是新鲜制备的CoB/CeO2催化剂样品放大5000 倍的SEM 图,将Co2+负载到载体CeO2上并常温化学还原后会形成CoB 纳米粒子,因此图2(b)显示CeO2载体表面形貌在经过钴离子浸渍和化学还原后,微观结构与纯CeO2载体有了明显不同,这是由于表面覆盖了CoB非晶粒子所致,从图中没有发现CoB 的严重团聚现象。图2(c)为CoB/CeO2-air(500℃)的微观形貌,与图2(b)相比,CoB/CeO2-air(500℃)催化剂中CeO2聚集成大团簇,该团簇紧密结合使表面微孔结构消失,说明高温空气焙烧会使CeO2粒子趋于聚集,特别是可以发现图2(c)中CeO2颗粒所具有的半空球结构坍塌,表面结构趋于收缩可能会使部分CoB 颗粒被包裹在CeO2内,不利于发挥CoB活性组分的催化活性。图2(d)为CoB/CeO2-N2(500℃)的微观形貌,相对于图2(c)的空气焙烧,在氮气中焙烧新鲜制备的CoB/CeO2催化剂会形成表面疏松多孔的蜂窝状结构,而且发现CoB 没有严重的团聚现象。

图2 不同催化剂样品的SEM图
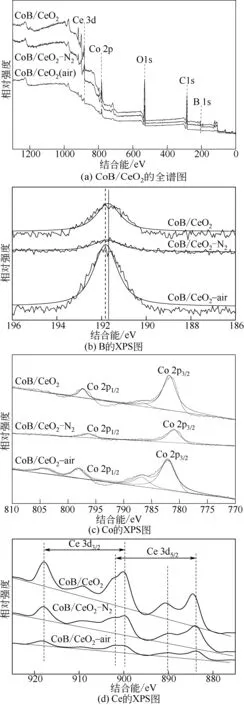
图3 不同CoB/CeO2 催化剂样品的XPS对比图
为了深入了解CoB/CeO2催化剂在不同焙烧处理气氛中的微观信息变化,本研究又对CoB/CeO2、CoB/CeO2-air(500℃)、CoB/CeO2-N2(500℃)进 行 了XPS分析,重点在于分析不同处理气氛中活性组分CoB 的化学价态等微观信息。图3(a)给出了3 种催化剂的全谱分析,从图中除了看出不同元素含量强度不同外,很难分析出三种催化剂的细微差别。图3(b)为3 种催化剂中硼元素的XPS 分析,可以看出新鲜制备的CoB/CeO2催化剂硼元素结合能大约在191.7eV,而经过空气焙烧以后硼的结合能明显向高能移动,其结合能大约为191.9eV,其结合能的增加说明硼元素很可能在空气焙烧气氛中发生了氧化。CoB/CeO2-N2(500℃)催化剂中硼结合能与新鲜制备催化剂类似,大约为191.7eV,说明在氮气焙烧气氛中硼仍然保持了与钴元素的有效结合。图3(c)给出了不同催化剂中钴元素的价态分布,可以知道在钴元素的XPS 谱图中,Co 2p3/2和Co 2p1/2自旋轨道峰分别位于778.0~792.0eV和794.0~797.0eV结合能区域。CoB/CeO2-air(500℃)催化剂中Co 2p3/2结合能大约为780.98eV,Co 2p1/2结合能为796.38eV,其自旋轨道分裂距离Δ(Co 2p1/2-Co 2p3/2)为15.5eV,该结果说明在CoB/CeO2-air(500℃)中表面钴元素为+3 价。对于新鲜催化剂CoB/CeO2和氮气气氛中焙烧的CoB/CeO2-N2(500℃)催化剂,其Co 2p3/2 的结合能增加到781.54eV 和781.96eV,并且在786.65eV附近都伴有较强的卫星峰,这说明在这两种催化剂中表面钴元素处于较低的价态,大约+2价或更低。在这两种催化剂中钴元素自旋轨道分裂距离Δ(Co 2p1/2-Co 2p3/2)分别为15.83eV 和16.06eV。这说明不同处理气氛会显著影响催化剂中CoB活性组分表面的氧化状态。图4(d)给出了Ce 元素的Ce 3d5/2和Ce 3d3/2谱,通过对比可以看出3 种样品在XPS 结合能位置上是一致的,说明Ce 的价态集中于四价,但是峰的强度明显不同,新鲜制备的CoB/CeO2和CoB/CeO2-N2(500℃)表面元素铈的分布状态基本一致,而在空气中焙烧得到的CoB/CeO2-air(500℃)催化剂表面铈元素分布有所下降,这可能是空气焙烧会改变CoB 分布从而诱导CeO2载体表面状态发生变化。
2.3 不同催化剂在NaBH4水解制氢中的催化性能
为了深入研究不同焙烧气氛对CoB/CeO2负载型催化剂活性的影响,3种不同方法制备的催化剂[新鲜合 成CoB/CeO2、CoB/CeO2-air(300℃/400℃/500℃)、CoB/CeO2-N2(300℃/400℃/500℃)]分别应用于NaBH4水解制氢。图4(a)为3种催化剂在焙烧温度为300℃时的催化活性对比情况,从图中可以看出CoB/CeO2-air(300℃)、CoB/CeO2-N2(300℃)展示了相近的催化活性,其制氢速率大约为2666.7mL/(min·g),都明显高于新鲜制备未经气氛焙烧的催化剂[1383.67mL/(min·g)]。当焙烧温度提高到400℃后,从图4(b)可以看出,CoB/CeO2-air(400℃)的催化活性略有升高,其制氢速率达到了2693.3mL/(min·g),而氮气中焙烧得到的CoB/CeO2-N2(400℃)催化活性却显著下降,但是仍然高于新鲜制备未经气氛焙烧的CoB/CeO2催化剂[1383.67mL/(min·g)]。当进一步提高催化剂焙烧温度达到500℃时,CoB/CeO2-air(500℃)的催化活性明显下降,不再具有NaBH4水解制氢催化活性。而CoB/CeO2-N2(500℃)催化剂仍然保持了较高的催化活性,前面分析CoB在氮气中焙烧会出现晶化,但是其较高的催化活性说明通过硼氢化钠反应体系的原位还原,CoB/CeO2-N2(500℃)催化剂仍然可以恢复较高的催化活性,并且CoB/CeO2在氮气气氛下焙烧能显著提高CoB活性组分与CeO2的连接强度,CoB在CeO2载体上保持了较高的分散度,因而催化活性较为稳定。而常温化学还原制备的CoB/CeO2催化剂,CoB与载体的相互作用较低,催化剂易于团聚,虽然初始活性较高,但是活性组分在使用中会逐步团聚,因而催化活性低于氮气焙烧处理的催化剂。在空气中高温处理CoB/CeO2催化剂虽然也会提高CoB 在CeO2载体的分散度和固载强度,但是更易造成CoB活性组分的永久性失活,且实验表明其催化活性无法通过硼氢化钠原位还原恢复。图4(c)对比了氮气气氛焙烧CoB/CeO2催化剂的催化活性随着处理温度的变化趋势,从图中可以看出随着焙烧温度的升高,其催化活性逐步升高,但是在400~500℃之间,其催化活性上升不明显,这可能与高温晶化有关。
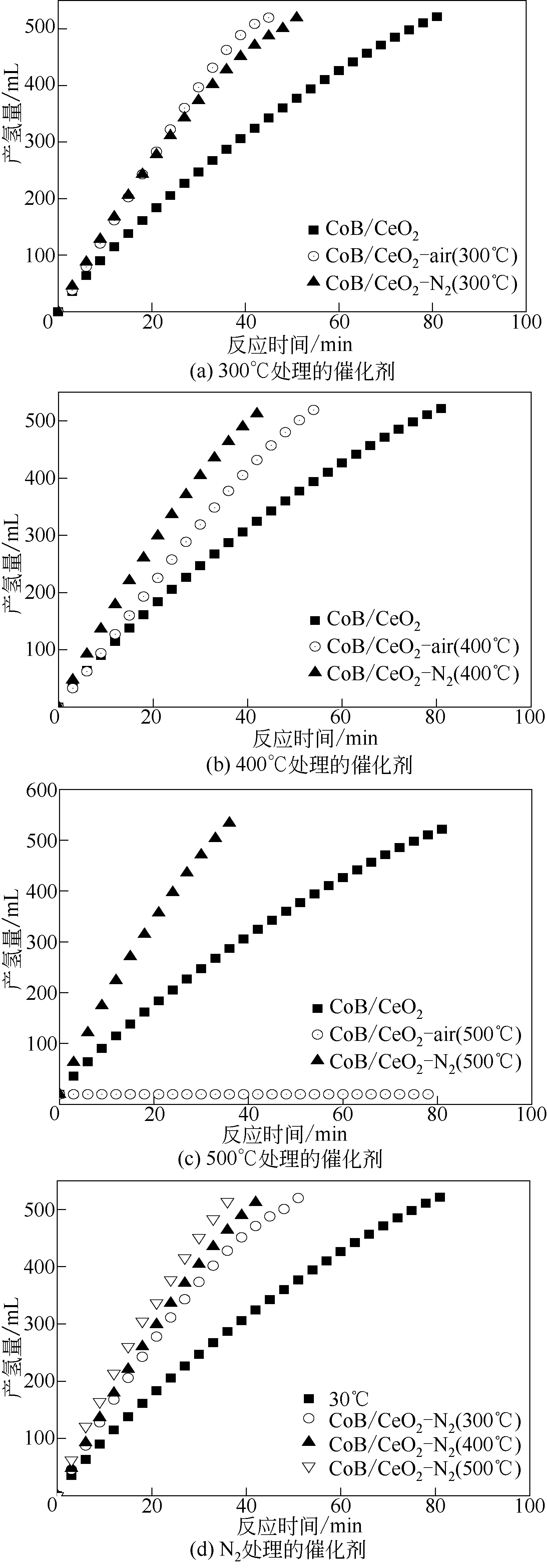
图4 不同处理温度下催化剂样品的催化活性
2.4 焙烧气氛对催化剂稳定性的影响
众所周知,CoB 催化剂虽然通常具有较高的催化活性,但是CoB 粒子尺度小、磁性强,CoB在使用过程中易于团聚,催化活性会逐步下降。通过CeO2载体负载会提高CoB 活性组分的分散度,能够抑制团聚。本研究对氮气焙烧处理的CoB/CeO2-N2(500℃)催化剂进行了循环稳定性测试。从图5(a)可以看出氮气气氛焙烧后的CoB/CeO2-N2(500℃)保持了较高的催化稳定性,例如第一次使用时,CoB/CeO2-N2(500℃)的初次使用制氢速率为2884mL/(min·g),第二次循环中其制氢速率略有下降[2622mL/(min·g)],第5 次循环中催化剂仍然保持了2149mL/(min·g)的制氢速率。对新鲜制备的CoB/CeO2(未经惰性气氛高温处理)催化剂进行循环稳定性测试,研究发现首次循环制氢速率为1418.2mL/(min·g),第二次使用制氢速率为1158mL/(min·g),第5 次循环制氢速率为574mL/(min·g),相对于首次循环其制氢速率下降了大约59.5%。对比上述两种催化剂可以看出,CoB/CeO2-N2(500℃)保持了更高的循环稳定性。
3 结论
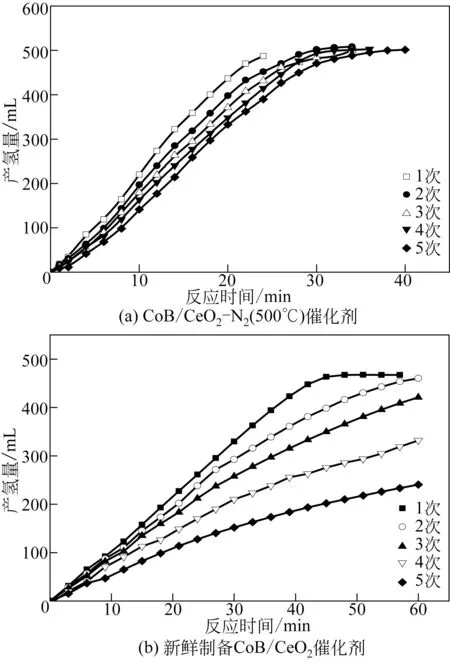
图5 CoB/CeO2催化剂的循环稳定性测试对比
本文首先采用溶胶-凝胶法制备了CeO2催化剂载体,进而通过化学还原法在CeO2表面负载了CoB 活性组分制备了负载型CoB/CeO2,系统研究了CoB/CeO2催化剂在不同焙烧气氛下的微观结构和催化活性。XRD 表征表明,负载在CeO2载体后催化剂无论在空气中焙烧还是氮气气氛下焙烧都仅仅显示了CeO2的萤石相结构,但对比分析表明其出现了晶化现象。XPS 分析表明CoB/CeO2、CoB/CeO2-N2和CoB/CeO2-air 这3 种催化剂中硼钴元素化学价态明显不同,CoB/CeO2-air 中硼钴在氧气中会趋于深度氧化。在硼氢化钠水解制氢反应中,CoB/CeO2-N2催化剂展示了更高的催化活性,并且随着焙烧温度的升高其催化剂活性略有提升。CoB/CeO2-air 催化剂在低温下焙烧(300℃)仍然具有一定的催化活性,而当焙烧温度提高到500℃, 其催化活性会完全丧失。CoB/CeO2-N2催化剂展示了较高的循环稳定性,在5 次循环后其催化活性下降了大约25%,而常规未经高温处理的CoB/CeO2催化剂催化活性会下降约60%,因此氮气焙烧后的负载型CoB/CeO2-N2催化剂具有更高的循环稳定性。
