蒙药制剂棘豆止咳散质量标准研究
2020-08-14扎拉嘎白乙拉吴凤娇郝俊生王青松呼格吉勒杨立国苏都那布其包晓华王秀兰乌力吉
扎拉嘎白乙拉 杨 楠 吴凤娇 郝俊生, 王青松 呼格吉勒 杨立国, 苏都那布其 包晓华, 王秀兰, 奥·乌力吉,
1.库伦旗蒙医医院,内蒙古 通辽 028000;2.内蒙古蒙医药工程技术研究院,内蒙古 通辽 028000;3.内蒙古民族大学蒙医药学院,内蒙古 通辽 028000
棘豆止咳散为内蒙古通辽市库伦旗蒙医医院院内制剂达格沙-4的优化改进处方,是由多叶棘豆、川贝母、甘草、石膏等4味蒙(中)药组成,该药对于咳嗽具有明显疗效,且价格低廉,无毒副作用,老人、儿童均可使用[1]。为了更好地控制棘豆止咳散质量,保证临床用药安全、有效,笔者对其进行质量标准研究,采用粉末显微鉴别方法对制剂中的多叶棘豆、川贝母、甘草和石膏进行了定性鉴别;采用薄层色谱法对制剂中川贝母和甘草进行了定性鉴别;根据2015年版《中华人民共和国药典》对其粒度、外观均匀度和水分进行了测定;通过HPLC法测定了其中甘草苷的含量。所建立的鉴别、检查和含量测定方法简便易行,重复性好,可用于生产过程中棘豆止咳散的质量控制,为临床上更加安全、可靠、有效的应用棘豆止咳散奠定了方法基础。现报道如下。
1 仪器与试剂
LC-20AT 高效液相色谱仪(岛津公司);Nikon E2000电子显微镜和DS-U3拍照装置(北京恒三江仪器销售公司);T-1700M马弗炉(郑州天纵电气设备公司);DHG-9023A型电热恒温箱(宁波江南仪器厂);BS224 S分析天平(北京赛多利斯仪器系统公司);FW-177高速万能粉碎机(天津市泰斯特仪器有限公司);HW-526数显恒温水浴锅(上海恒科学仪器有限公司);硅胶G(薄层色谱)、硅胶GF254(薄层色谱),青岛海洋化工有限公司制造;薄层色谱硅胶G板(批号20170902),青岛海洋化工厂产;薄层色谱硅胶GF254板(批号20170608),青岛海洋化工厂产;棘豆止咳散(库伦旗蒙医医院自制,批号201902001-201902003);实验用多叶棘豆药材由赤峰荣兴堂药业有限责任公司蒙药饮片厂提供(生产批号:1807046);甘草药材由赤峰荣兴堂药业有限责任公司蒙药饮片厂提供(生产批号:170628);川贝母药材由赤峰荣兴堂药业有限责任公司蒙药饮片厂提供(生产批号:1701036);生石膏药材由赤峰荣兴堂药业有限责任公司蒙药饮片厂提供(生产批号:1703067);甘草苷对照品(生产批号:111610-201607,中国食品药品检定研究院);阴性对照样品(内蒙古蒙医药工程技术研究院自制);95%乙醇(天津市永大化学有限公司);乙腈为色谱纯,水为超纯水,其他试剂均为分析纯。
2 方法与结果
2.1 棘豆止咳散中多叶棘豆、川贝母、甘草、石膏的显微鉴别 根据显微鉴别方法(《中国药典》2015年版四部通则2001)试验。取棘豆止咳散(201902001)适量,水合氯醛、稀甘油制片,放显微镜下观察,结果如图1所示。

图1 棘豆止咳散显微特征图
2.1.1 多叶棘豆 纤维多成束,碎断,淡黄色,壁较厚,直径10~40 μm;非腺毛为单细胞,长约88~430 μm,直径15~50 μm;叶上、下表皮细胞为长方形或不规则形,垂直壁弯曲,并有不定式气孔,类圆形或椭圆形,上表皮气孔略小。
2.1.2 川贝母 淀粉粒众多,呈广卵形、贝壳形、肾形或椭圆形,直径40~60 μm,脐点短缝状、人字状或马蹄状,层纹可察见。螺纹导管和网纹导管直径可达64 μm。
2.1.3 甘草 粉末淡棕黄色。纤维成束,直径8~15 μm,壁厚,纤维束周围薄壁细胞含草酸钙方晶,形成晶纤维。草酸钙方晶多见。具缘纹孔导管较大,稀有网纹导管。木栓细胞红棕色,多角形。
2.1.4 石膏 不规则片状无色结晶,有平直纹理。低负突起,糙面不显著。
2.2 棘豆止咳散中川贝母、甘草的薄层鉴别
2.2.1 棘豆止咳散中川贝母的薄层鉴别 取3批次棘豆止咳散各12 g,加氨试液25 mL,密塞,浸泡1 h,加二氯甲烷25 mL,超声处理1 h,滤过,滤液用3%盐酸振摇提取3次,每次8 mL,合并酸液,用氨水调pH=10,用二氯甲烷振摇提取3次,每次10 mL,合并二氯甲烷提取液,水浴蒸干,残渣加1 mL甲醇使溶解,作为供试品溶液。另取川贝母对照药材1 g,同法制成川贝母对照药材溶液。再取多叶棘豆药材2 g、甘草药材2 g、石膏1 g,分别打粉、过筛、混合均匀,同法制成阴性溶液。照薄层色谱法(中国药典2015年版四部通则0502)试验,吸取上述5种溶液各3 μL,分别点于同一硅胶G薄层板上,以乙酸乙酯-甲醇-氨水-水(25∶1∶1∶0.1)为展开剂,展开,取出,晾干,依次喷稀碘化铋钾试液和亚硝酸钠乙醇试液。结果如图2所示。
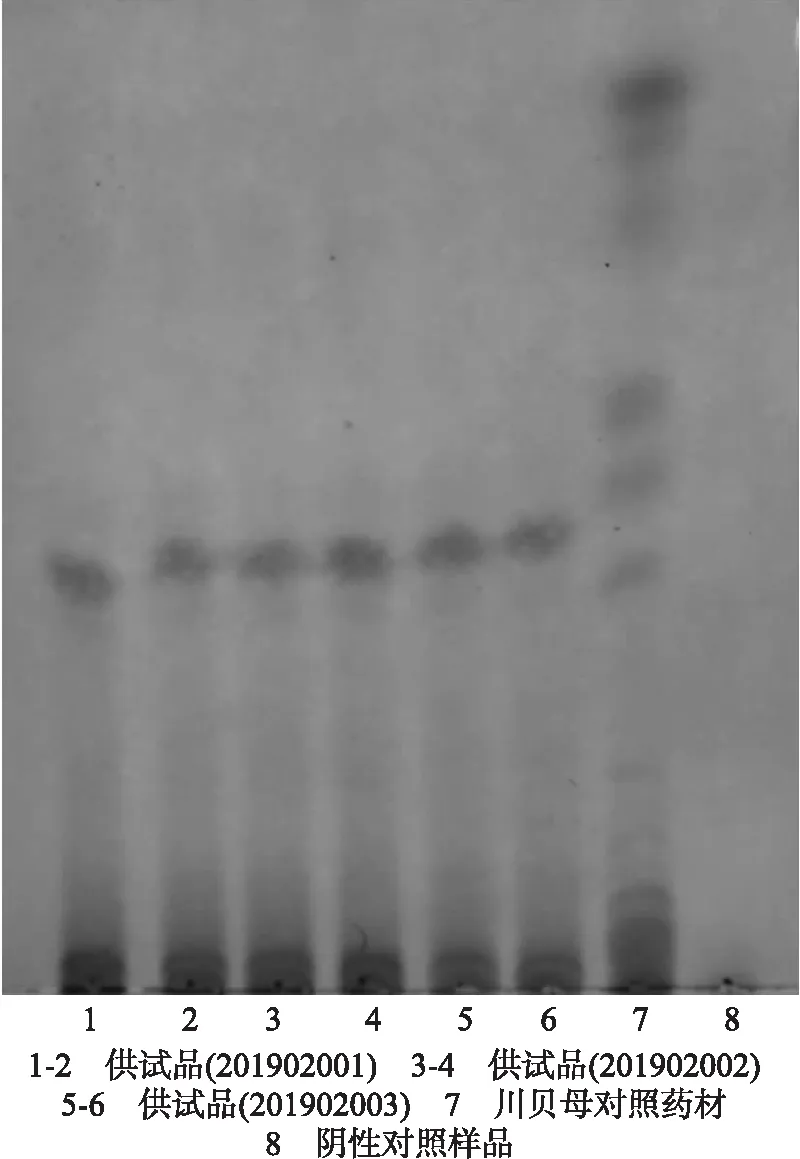
图2 棘豆止咳散中川贝母薄层鉴别色谱图
2.2.2 棘豆止咳散中甘草的薄层鉴别 取3批次棘豆止咳散各1 g,加65%乙醇10 mL,加热回流2 h,滤过,滤液蒸干,残渣加甲醇1 mL使溶解,作为供试品溶液。另取甘草对照药材1 g,同法制成对照药材溶液。再取甘草苷对照品,加甲醇制成每1 mL含0.5 mg的溶液,作为对照品溶液。照薄层色谱法(中国药典2015年版四部通则0502)试验,吸取上述5种溶液各3 μL,分别点于同一硅胶G薄层板上,以乙酸乙酯-甲酸-冰醋酸-水(15∶1∶1∶2)为展开剂,展开,取出,晾干,喷以10%硫酸乙醇溶液,在105 ℃加热至斑点清晰。结果如图3所示。
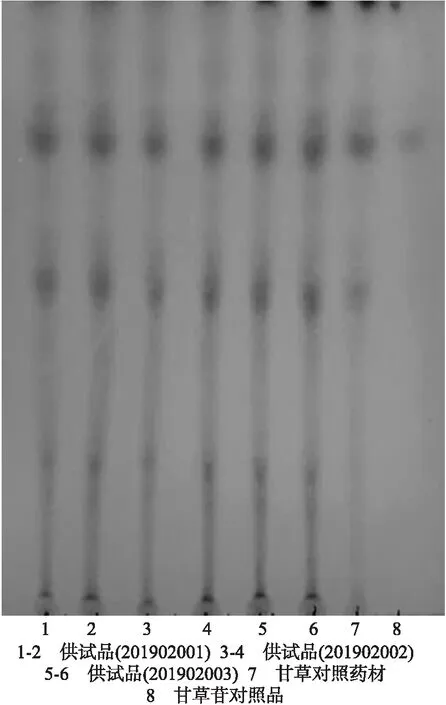
图3 棘豆止咳散中甘草薄层鉴别色谱图
2.3 棘豆止咳散粒度、外观均匀度和水分测定 根据2015年版《中华人民共和国药典》四部通则0982、0115、0832测定三批次棘豆止咳散中粒度、外观均匀度、水分进行了测定,结果见图4、表1。

图4 棘豆止咳散外观均匀度检查

表1 棘豆止咳散粒度、水分测定结果
2.4 棘豆止咳散中甘草苷含量测定
2.4.1 色谱条件 色谱柱:X Terra®MSC18 5.0 μm (4.6 mm×250 mm);流动性:A相为乙腈,B相为0.05%磷酸水溶液;按表2进行梯度洗脱;检测波长237 nm;柱温40 ℃;流速1.0 mL·min-1;检测时间40 min;进样量10 μL。在上述条件下,供试品溶液中甘草苷色谱峰与其他相关峰均能达到较好分离,分离度大于1.5,理论塔板数以甘草苷计不低于2000。按棘豆止咳散处方制备工艺自制不含甘草药材的阴性供试品溶液,按上述色谱条件进样,结果阴性溶液对样品测定无干扰。结果如图5所示。

表2 流动相梯度洗脱表
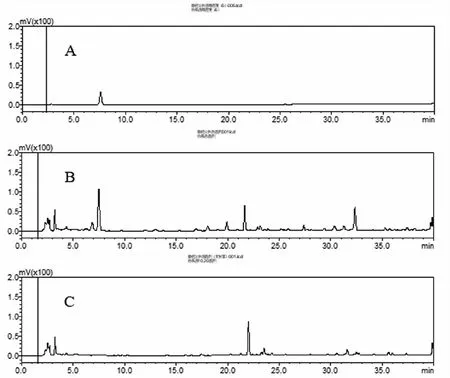
图5 甘草苷(A)、棘豆止咳散(201902001)、阴性对照样品的HPLC色谱图
2.4.2 对照品和供试品溶液的制备
2.4.2.1 对照品溶液的制备 取甘草苷对照品,精密称定,加70%乙醇制成每1 mL含甘草苷0.1 mg的溶液,备用。
2.4.2.2 供试品溶液的制备 取棘豆止咳散约0.5 g,精密称定,置具塞锥形瓶中,精密加入50%甲醇30 mL,密塞,称定重量,超声处理30 min,放冷,再称定重量,用50%甲醇补足减失的重量,摇匀,过滤,取续滤液,备用。
2.4.3 线性关系考察 精密称量甘草苷对照品1.08 mg于10 mL容量瓶中,加70%乙醇溶液使其溶解,并稀释至刻度,得其甘草苷浓度为0.108 mg/mL。分别精密吸取5、7.5、10、12.5、15μL,注入高效液相色谱仪,按“2.4.1”项下条件进行测定,以对照品色谱峰面积为纵坐标,进样量为横坐标,绘制标准曲线。回归方程为y=198188x+184557,其中r2=0.9993,说明甘草苷在上述范围内有良好线性关系。
2.4.4 精密度试验 精密吸取“2.4.2”项下对照品溶液,连续进样6针,按“2.1”项下色谱条件进行测定,根据其峰面积计算其RSD值为0.502%,表明该仪器精密度良好。
2.4.5 重复性试验 精密称量同一批次棘豆止咳散粉末6份,分别按照“2.4.2”供试品溶液制备方法制备,按“2.4.1”项下色谱条件进行测定,同一天内连续进样6针,根据其峰面积计算其RSD值为0.632%,表明方法重复性良好。
2.4.6 稳定性试验 按“2.4.1”项下色谱条件进行测定,精密称取同一批棘豆止咳散散剂粉末一份,按照“2.4.2”供试品溶液制备方法制备,分别于0、2、4、8、12、24 h进样,根据其峰面积计算其RSD值为2.849%,表明供试品溶液在24 h内稳定。
2.4.7 回收率试验 精密称量甘草苷对照品10.06 mg于10 mL容量瓶中,加70%乙醇溶液使其溶解,并稀释至刻度,得其甘草苷浓度为1.006 mg/mL。精密称取同一批棘豆止咳散粉末9份,每份0.25 g,在其中加入甘草苷对照品0.5 mL、1 mL、1.5 mL,每组三份,共九份。按照“2.4.2”供试品溶液制备方法制备,按“2.4.1”项下色谱条件,结果见表3,根据其回收率计算其RSD值为2.968%。
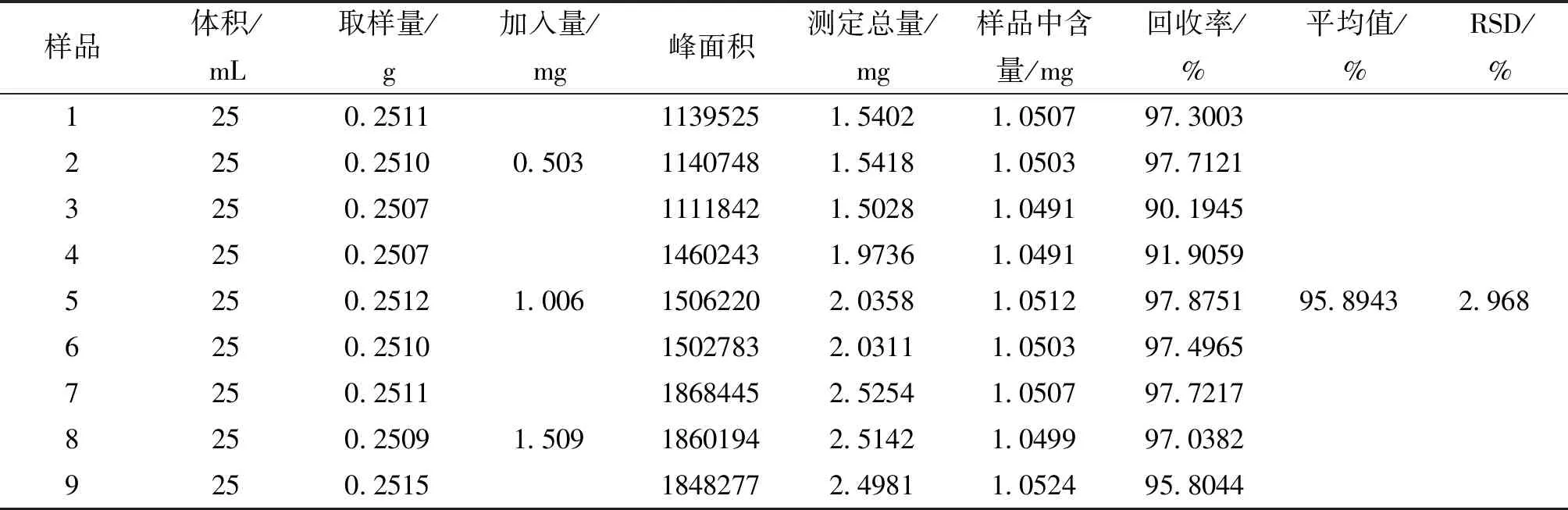
表3 回收率试验结果
2.4.8 样品测定 分别精密吸取对照品溶液和三批次供试品溶液各10μL,注入高效液相色谱仪测定,计算得到三批次棘豆止咳散中甘草苷的含量见表4。

表4 棘豆止咳散甘草苷含量测定结果
3 讨论
3.1 定性鉴别 棘豆止咳散中川贝母、甘草、石膏均为2015版《中国药典》收载药材[2],粉末显微特征明显[3-5],参照2015版《中国药典》方法对上述三味药材进行了鉴别;多叶棘豆为1998版中华人民共和国卫生部药品标准-蒙药分册收载药材[6],但其中未见显微鉴别内容,我们参照相关文献[7-8]对多叶棘豆进行了显微鉴别。
多叶棘豆为本处方君药,但由于多叶棘豆药材化学成分较为复杂[9-13],主产地不明确,不同批次多叶棘豆化学成分差别较大,因此,本研究未能确定处方中源自多叶棘豆的主要指标成分,加之市场上尚无法定的多叶棘豆对照药材,所以,本研究薄层鉴别中仅对处方中川贝母和甘草进行了薄层鉴别,未能对多叶棘豆进行薄层鉴别。
3.2 定量测定 在参照相关文献基础上[14-15],本研究比较了甲醇-水、乙腈-水系统,发现以乙腈-水为流动相,检测波长为237 nm时,被测组分的分离效果好,分离度符合要求。从2.4.3可知,相关系数r2=0.9993,说明甘草苷在上述范围内有良好线性关系。其精密度、稳定性、重复性的RSD值均小于3.0%,从表3可以看出,其回收率在90.19%~97.87%,满足中国药典规定的当待测组分含量为0.1%时,其回收率限度为90%~108%的要求。
4 结论
本研究建立了棘豆止咳散中各组方药材显微鉴别方法、棘豆止咳散中川贝母和甘草的薄层鉴别方法及棘豆止咳散中甘草苷的含量测定方法,试验表明以上方法操作简单,灵敏度高,重复性好,专属性强,可作为棘豆止咳散的质量控制方法,为临床上更加安全、可靠、有效的应用棘豆止咳散奠定方法基础。
