基于16SrRNA基因高通量测序方法分析多花黄精内生细菌群落结构及多样性
2020-08-12蔡媛刘浩王勇庆谢景黄建华劳嘉贺炜张水寒
蔡媛 刘浩 王勇庆 谢景 黄建华 劳嘉 贺炜 张水寒


〔摘要〕 目的 采用高通量测序技术分析多花黄精内生细菌的多样性及菌群结构。方法 通过蒽酮-硫酸法测定多糖含量,采用通用引物对细菌16S rRNA高可变区(V3-V4区)进行PCR扩增,运用Illumina Miseq PE250高通量測序技术对扩增子进行测序,并用QIIME等软件对测序序列进行生物信息学分析。通过Spearman相关性分析菌群与多糖含量的相关性。结果 多花黄精内生菌测序共获得90 628条有效序列和5 287个OTU,稀释曲线和Coverage指数反映测序结果比较全面地覆盖了多花黄精内生细菌群落。Alpha多样性分析表明,其内生细菌多样性程度高。在门水平,其优势菌门为变形菌门(Proteobacteria)、放线菌门(Actinobacteria)、芽单胞菌门(Gemmatimonadetes)、厚壁菌门(Firmicutes)、酸杆菌门(Acidobacteria)。属水平,核心菌群由鞘氨醇单胞菌属(Sphingomonas)、芽单胞菌属(Gemmatimonas)、链霉菌属(Streptomyces)组成。PICRUSt基因预测表明,多花黄精内生细菌以代谢功能为主,主要包括能量代谢、碳水化合物代谢、氨基酸代谢等。通过Spearman相关性分析,在属水平共得到19个菌群与多糖含量相关,其中3个属呈正相关,16个属菌群呈负相关。结论 多花黄精内生细菌的多样性程度高,存在多种不同种类的细菌,且菌群与多糖含量相关。该研究解析了多花黄精内生细菌的多样性、丰度及主要菌属,对深入研究多花黄精内环境有一定指导意义。
〔关键词〕 多花黄精;16S rRNA;内生细菌;多样性;群落结构
〔中图分类号〕R284.1 〔文献标志码〕A 〔文章编号〕doi:10.3969/j.issn.1674-070X.2020.07.013
〔Abstract〕 Objective To analyze the bacterial diversity and their community structure from Polygonatum cyrtonema using high-throughput sequencing technology. Methods The polysaccharide content was determined by anthrone-sulfuric acid method. PCR amplification of 16S rRNA high variable region (V3/V4 region) of bacteria was performed by using universal primers. The sequencing of amplicons was conducted by using Illumina Miseq PE250 high-throughput sequencing technology, and bioinformatics analysis of sequencing sequences was performed using software such as QIIME. The correlation between endophytic bacteria and polysaccharide was analyzed by spearman correlation. Results A total of 90 628 effective sequences and 5 287 OTUs were obtained from the sequencing of endophytic bacteria in Polygonatum cyrtonema, and the dilution curve and coverage index reflected that the sequencing results comprehensively covered the endophytic bacterial community of Polygonatum cyrtonema. Alpha diversity analysis showed that the endophytic bacteria were highly diverse. At the level of the phylum, the dominant bacteria were Proteobacteria, Actinobacteria, Gemmatimonadetes, Firmicutes, and Acidobacteria. At the genus level, the core flora consists of Sphingomonas, Gemmatimonas, and Streptomyces. The PICRUSt gene prediction indicated that endophytic bacteria of Polygonatum cyrtonema were mainly metabolized, including energy metabolism, carbohydrate metabolism, and amino acid metabolism. Through spearman correlation analysis, the polysaccharide content associated with endophytic bacteria were obtained 19 bacterias at genus level. Among them, three genera were positively correlated and 16 genera were negatively correlated. Conclusion The diversity of endophytic bacteria in Polygonatum cyrtonema is high. There are many different kinds of bacteria, and the flora is related to polysaccharide content. The study analyzed the diversity, abundance and main genus of endophytic bacteria of Polygonatum cyrtonema, which has certain guiding significance for deep study on the internal environment of Polygonatum cyrtonema to quality.
〔Keywords〕 Polygonatum cyrtonema; 16S rRNA; endophytic bacteria; diversity; community structure
多花黄精为百合科(Liliaceae)黄精属(Polygonatum Mill.)多年生草本植物,以其根茎入药,在我国拥有悠久的药用历史,具有补气养阴、健脾、润肺、益肾等功效[1]。多花黄精主要活性成分为黄精多糖,还含有薯蓣皂苷元、黄酮、氨基酸等成分,具有增强免疫力、抗病毒、延缓衰老等作用,是一味具有广阔开发前景的药食同源类中药材[2-3]。
植物内生菌是指在其生活史的某一个阶段或整个阶段生活于健康植物各种组织和器官内部或细胞间隙并且正常情况下不引起病害的微生物。内生菌广泛分布于低等植物和高等植物的各个组织中,经长期协同进化,内生菌可帮助宿主合成或独自合成对宿主植物有利的活性成分[4]。研究者发现陈皮在陈化过程中,其表面微生物可以引起陈皮药效物质成分变化,证明微生物菌群参与了陈皮中药材品质的变化[5-6]。目前,关于多花黄精菌群结构的分析未见报道,故本文首次研究了多花黄精根茎内生菌群的结构组成,为后续探讨内生菌对多花黄精品种的影响奠定基础。
既往对植物内生菌的分离鉴定和生物學特征分析都是基于纯培养,但微生物种类繁多,因此采用纯培养方式对植物内生菌进行正确分类存在较大困难,通过传统的、依赖于培养的分离方法鉴定的微生物只占植物内生菌总数的0.1%~10%,难以明确整体微生物菌群群落结构。第二代高通量测序技术对微生物进行深度测序,精确检测低至万分之一的痕量微生物,突破了传统方法基于纯培养的限制,可客观而真实反映微生物群落自身及其与宿主之间的相互作用关系[7-9]。高通量测序不仅能确定样品中微生物群落组成,还能明确其相对含量,为植物样本中的内生菌微生物群落的多样性分析提供一种可靠的有效的方法。目前,高通量测序技术在土壤根际微生物、植物内生菌、肠道菌群等样品中的微生物多样性分析得到广泛应用[10-12]。故本研究通过基于Illumina MiSeq平台的第二代测序技术对多花黄精内生细菌的种类进行多样性组成谱测序,并对其内生细菌多样性及其群落结构进行差异分析,旨在明确多花黄精根茎中的优势及特有菌群,并初步分析菌群与多糖含量之间的相关性,为后期深入探究多花黄精的品质及功能菌群的寻找研究提供科学依据。
1 材料
1.1 样品
多花黄精采于湖南省新化县野生林下(111°05'37.25″E, 27°40'59.05″N),经湖南省中医药研究院生药室刘浩主任鉴定为百合科黄精属植物多花黄精(Polygonatum cyrtonema Hua),采集后去除泥沙分成两份,一份保存于-80 ℃冰箱,一份烘干用于多糖含量测定。
1.2 主要仪器和试剂
UV 2500紫外-可见分光光度计(上海第三分析仪器厂);NanoDrop One核酸蛋白测定仪(ND-2000);DNA凝胶电泳仪(北京六一DYCP-31DN);PCR扩增仪(德国Eppendorf AG 22331);植物DNA提取试剂盒(Omega公司);高效高保真酶(NEB公司);核酸纯化试剂盒(Axygen公司)。
2 方法
2.1 总DNA提取
取鲜多花黄精的根茎用流水冲洗30 min,去除表面泥沙后于超净工作台内用75%乙醇中浸泡5 min,接着置于8%次氯酸钠浸泡2 min,最后用0.1%的氯化汞浸漂4 min,用无菌磷酸盐缓冲溶液清洗多次获得表面无菌的多花黄精。将多花黄精切成两半,从中间掏取多花黄精块,然后切成小块并用无菌磷酸盐缓冲溶液反复冲洗,取冲洗液于10 000 r/min 离心10 min,倒掉上清液,合并沉淀用于DNA提取。参照DNA提取试剂盒说明书提取总DNA,琼脂糖凝胶电泳检测提取DNA的纯度。
2.2 PCR扩增及测序
总DNA用Nanodrop进行精确定量。PCR反应体系所用的引物为细菌16S rRNA V3-V4区通用引物338F: 5'-ACTCCTACGGGAGGCAGCA-3'和806R: 5'-GGACTACHVGGGTWTCTAAT-3'。PCR反应体系(50 μL):gDNA(50 ng/μL)1.0 μL,上、下游引物(10 μmol/L)各1.25 μL,高保真酶(5 U/μL)0.25 μL, dNTPs(10 mmol/L)1.0 μL,10×buffer 5.0 μL,ddH2O 40.25 μL。PCR反应条件:98 ℃ 5 min;98 ℃30 s,52 ℃ 30 s,72 ℃ 1 min,25个循环;72 ℃ 5 min。PCR产物用琼脂糖凝胶电泳检测,并用核酸纯化试剂盒纯化回收。采用Quan-iT PicoGreendsDNA Assay Kit检测试剂盒对回收产物精确定量,将样品DNA浓度调整为20 pmol/L上机测序。测序委托上海派森诺生物科技股份有限公司完成,每个样本3个重复。
2.3 多花黄精多糖含量测定
采用蒽酮-硫酸法测定多糖含量,具体参考《中华人民共和国药典》2015年版。
2.4 数据处理及分析
2.4.1 原始测序数据处理 采用滑动窗口方法对FASTQ格式双端序列进行质量筛查,要求窗口中碱基平均测序准确率≥99%。随后利用FLASH软件对通过质量初筛的双端序列根据重叠碱基进行配对连接[13],根据样本所对应的Index信息,将连接后的序列与样本完全匹配,得到每个样本最终的有效数据。
2.4.2 物种注释及多样性分析 通过QIIME软件调用UCLUST序列比对工具[14],默认以97%的序列相似度进行归并和操作分类单元(operational taxonomic units, OTU)划分,选取相对丰度值最高的OTU代表序列与Unit数据库进行比对和物种注释,并统计样品在各个分类水平门、纲、目、科、属的丰度信息。使用QIIME软件分析计算样品的Alpha多样性值,包括Shannon指数、Chao 1指数、Simpson指数、ACE指数。使用MEGAN软件构建样品聚类树,PICRUSt基因预测内生细菌的生物学功能,综上数据统计分析得到多花黄精内生细菌群落结构组成。
2.4.3 多糖含量与菌群相关性分析 选取属水平总丰度前30的物种与多糖含量数据,通过Spearman相关系数,分析菌群与多糖含量之间的相关性。
3 结果
3.1 测序数据统计及多样性分析
多花黄精样品经Illumina MiSeq高通量测序分析,样品测得的序列长度分布在152~531 bp之间,通过质控优化共得到序列94 524条,序列平均长度409 bp。从序列长度的分布来看,与16S rRNA V3-V4区序列长度大致吻合。经与数据库比对分析,共有90 628条序列与数据库中的序列相似,序列有效性为95.88%,说明测序合理,可用于进一步分析。
对得到的多花黄精样品有效序列进行Shannon稀释曲线和等级聚类曲线分析。从Shannon曲线和等级聚类曲线(图1)可以看出,曲线已较平缓,说明测序趋于饱和,表明取样合理,通过对细菌16S rRNA V3-V4区的PCR扩增序列基本上能真实反映多花黄精内生细菌的群落组成。
Alpha多样性是指对样品中微生物群落物种多样性的分析,包括侧重于体现群落丰富度的ACE指数和Chao 1指数,以及兼顾群落均匀度的Simpson指数和Shannon指数。Alpha多样性指数见表1。Alpha指数常用于对比性分析,一般而言,ACE或Chao指数值越大,说明群落的丰富度越高。Simpsonn或Shanno指数值越高,说明群落的多样性越高,其值越低则说明物种的多样性越低。
3.2 OTU聚类统计及物种注释
多花黄精样品共获得94 524条过滤后得到的拼接序列,可分为5 287个OTU。使用数据库对OTU序列进行相似性比对,发现多花黄精内生菌分类学地位明确的细菌有20个门、48个纲、121个目、208个科、432个属。根据样品物种注释结果,选取在门、纲、目、科、属水平上最大丰度排名前10物种进行统计分析(如图2所示),在门水平上,Proteobacteria 和Actinobacteria分别占到了38.7%和16.6%,在纲的水平上,Gammaproteobacteria、Alphaproteobacteria和Gemmatimonadetes三者合计占比49.02%,在目水平上Gemmatimonadales和Betaproteobacteriales占比22.3%,在科的水平上,Gemmatimonadaceae和Burkholderiaceae合计占比21.52%,在属的水平上,排名前10的菌主要是Sphingomonas、Gemmatimonas、Streptomyces、Paucibacter、Thermoactinomyces、Massilia、Shigella、Bradyrhizobium、Cupriavidus、Acinetobacter。其中Sphingomonas是优势菌群,大约占7%。
3.3 样品中内生细菌物种类群组成分析
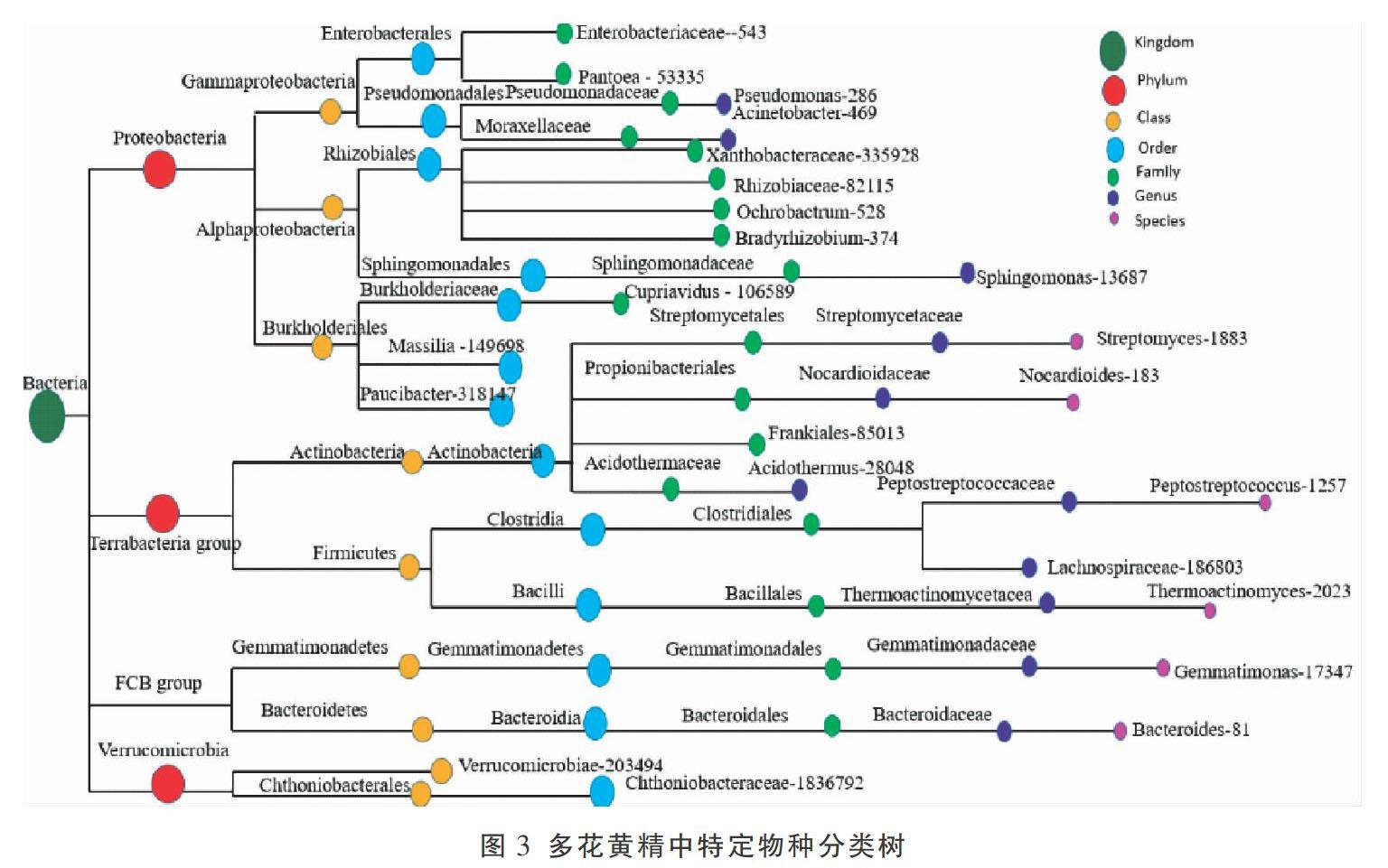
从样品中筛选相对丰度值前20进行物种分类树统计,样品的物种分类树见图3。从图3可以看出,多花黄精内生细菌种类主要在Proteobacteria、Verrucomicrobia、Terrabacteria group、FCB group 4个门,其中Proteobacteria占比为18%;从纲分类水平来看,主要是Gammaproteobacteria、Alphaproteobacteria、Burkholderiales、Actinobacteria、Firmicutes、Gemmatimonadetes、Bacteroidetes、Chthoniobacterales,其中Gammaproteobacteria和Alphaproteobacteria为优势菌纲;从目水平来看,Rhizobiales为优势菌目,占比达到了38.6%。从科分类水平来看,其优势菌群主要为Enterobacteriaceae、Pantoea、Gemmatinonadales等,其中Gemmatinonadales为优势菌科,占比达到了42.88%。从属水平来看,其优势菌群主要集中在Pseudomonas、Streptomycetaceae、Gemmatimonadaceae等,其中Gemmatimonadaceae为优势菌属,占比达到了50.16%。而基于16S rRNA二代测序水平注释到细菌种的序列很少,可以确定细菌种名的更少,大部分都无法定种。
3.4 物种系统进化关系分析
为深入分析多花黄精中内生细菌的系统进化关系,选取属水平所对应OTU相对丰度排名前10位的系统进化数据,结合每个OTU相对丰度及物种注释置信度进行整合,构建属水平OTU的系统进化关系(见图4)。该系统进化关系图分为两个部分,最里层为OTU代表序列构建的系统发育树,不同颜色代表其對应的属名;外层为OTU相对丰度值大小。从图中可以看出Paucibacter、Escherichia-Shigella、Cupriavidus、Massilia在系统进化上较为接近,聚为一支,而且其外层柱高差异性不明显,从属水平来看,这些物种的丰度差别不大,在进化上基本趋于一致。
3.5 内生细菌中的基因功能分类分析
通过对多花黄精内生细菌进行功能分类分析,共获得20类主要功能,具体如图5所示,主要体现在膜运输、代谢、细胞信号传导等方面。其中代谢包括能量代谢、碳水化合物代谢、氨基酸代谢、核苷酸代谢、脂质代谢、萜类及黄酮类化合物代谢、聚糖生物合成代谢等。上述获得的多花黄精内生细菌相关功能信息,为研究多花黄精内生细菌与其有效成分的生物合成提供了参考依据。
3.6 内生细菌与多糖含量相关性分析
通過蒽酮-硫酸法测得3个新化多花黄精样品多糖含量分别为5.3%、4.5%、8.2%,选取属水平总丰度前30的物种,通过Spearman相关系数分析,P<0.05,共得到19个与多糖含量相关的菌群,具体如图6所示,其中Actinobacteraia门中的Pseudomonas、Enterobacter、Brucella 3个属的菌群与多糖含量为正相关,表示该类菌群菌群丰度值越高,多花黄精中多糖含量越高。其余16个菌群与多糖含量为负相关,表示该类菌群菌群丰度值越高,多花黄精中多糖含量越低。
4 讨论
本研究首次利用高通量测序技术对多花黄精内生细菌进行研究,通过对16S rRNA基因的V3-V4高变区测序分析,检测多花黄精内生细菌群落结构多样性。样品共获得涵盖20个门、48个纲、121个目、208个科、432个属的细菌,表明多花黄精内生环境中存在着丰富的微生物,这些微生物不仅数量众多,且种类也很丰富。
在门水平上得到的优势菌群为变形菌门、放线菌门、芽单胞菌门,其中变形杆菌门丰度最高。研究表明,变形菌和放线菌有助于药用植物产量及有效物质含量的提高[15-16],在促植物生长,防治病害方面发挥着重要作用[17]。在属水平上,含量最高的为未分类细菌,这说明多花黄精样品中蕴含大量未知微生物种类,有关多花黄精内生细菌在属水平上的研究还不够深入,需要采用其他分析手段进一步鉴定确认。此外,分类学地位明确的、丰度居前10的菌主要是Sphingomonas、Gemmatimonas、Streptomyces、Paucibacter、Thermoactinomyces、Massilia、Escherichia-Shigella、Bradyrhizobium、Cupriavidus、Acinetobacter,这些菌群功能主要体现在降解酚类物质[18]、固氮解磷促生长[19-22]、土壤改良[23]、合成多种次级代谢产物和酶[24]、植物病变等方面[25-26],后续可依据其优势菌群功能对多花黄精进行深入研究,为解决多花黄精病变及有效物质分子形成机制提供技术支持。选取属水平丰度前30的物种与多糖含量进行相关性分析,共获得19个菌群与多糖含量呈显著相关。Pseudomonas、Enterobacter、Brucella与多糖含量呈显著性正相关,Sphingomonas、Gemmatimonas、Streptomyces、Paucibacter、Thermoactinomyces等16个菌群与多糖含量呈负相关。通过比对分析发现,优势菌群占比最大的,大部分与多糖含量呈负相关性,可能与内生细菌以多糖等碳水化合物为基础代谢底物进行代谢消耗有关。
本研究结果初步探讨了多花黄精内生细菌菌群结构多样性及其潜在功能特征,下一步的工作将重点从以下几个方面展开:(1)分离多花黄精根茎内生细菌菌株,评估其抗菌、抗虫害、产生多糖或黄酮等活性成分能力,筛选获得具有药用和工业应用潜力的菌株。(2)对不同生长环境、产地多花黄精的品质及其内生细菌进行分析,阐述其“菌-质”关联性。
参考文献
[1] 国家药典委员会.中华人民共和国药典:一部[S].北京:中国医药科技出版社,2015:306-307.
[2] 陈 辉,冯珊珊,孙彦君,等.3种药用黄精的化学成分及药理活性研究进展[J].中草药,2015,46(15):2329-2338.
[3] 姜程曦,张铁军,陈常青,等.黄精的研究进展及其质量标志物的预测分析[J].中草药,2017,48(1):1-16.
[4] HASSANI M A, DURáN P, HACQUARD S. Microbial interactions within the plant holobiont[J]. Microbiome, 2018, 6(1):58-75.
[5] 王 福,张 鑫,卢俊宇,等.陈皮“陈久者良”之黄酮类成分增加原因探究[J].中国中药杂志,2015,40(24):4890-4896.
[6] 张 鑫,刘素娟,王智磊,等.橘皮表面真菌群落结构多样性分析[J]. 微生物学通报,2017,44(5):1089-1098.
[7] SHENDURE J, JI H. Next-generation DNA sequencing[J]. Nature Biotechnology, 2008, 26: 1135-1145.
[8] MEDINGER R, NOLTE V, PANDEY R V, et al. Diversity in a hidden world: potential and limitation of next-generation sequencing for surveys of molecular diversity of eukaryotic microorganisms[J]. Molecular Ecology, 2010, 19(S1): 32-40.
[9] JAUFEERALLY-FAKIM Y. Assessing biodiversity: A molecular approach using DNA sequencing[M]. Novel Plant Bioresources, 2014, 69-80.
[10] FITZPATRICK C R, COPELAND J, WANG P W, et al. Assembly and ecological function of the root microbiome across angiosperm plant species[J]. Proceedings of the National Academy of Sciences, 2018, 115(6): 1157-1165.
[11] DONG L, JIANG X, ZHANG L, et al. Rhizospheric microbial communities are driven by Panax ginseng at different growth stages and biocontrol bacteria alleviates replanting mortality[J]. Acta Pharmaceutica Sinica B, 2018, 8(2):272-282.
[12] 王 艷,常 帆,程虎印,等.重楼根际及药用部位内生真菌多样性与群落结构差异分析[J].中草药,2019,50(5):1232-1237.
[13] MAGO T, SALZBERG S L. FLASH: fast length adjustment of short reads to improve genome assemblies[J]. Bioinformatics, 2011, 27(21): 2957-2963.
[14] EDGAR R C: Search and clustering orders of magnitude faster than BLAST[J]. Bioinformatics, 2010, 26(19): 2460-2461.
[15] YI X Y, WAN L D, YONG L. Characterization of soil bacterial communities in rhizospheric and nonrhizospheric soil of Panax ginseng[J]. Biochemical Genetics, 2012, 50(11-12): 848-859.
[16] JIN H, YANG X Y, YAN Z Q, et al. Characterization of rhizosphere and endophytic bacterial communities from leaves, stems and roots of medicinal Stellera chamaejasme L[J]. Systematic & Applied Microbiology, 2014, 37(5): 376-385.
[17] ZHANG P, QIN S, YUAN B, et al. Diversity and bioactivity of actinomycetes isolated from medicinal plant Taxus chinensis and rhizospheric soil[J]. Acta Microbiologica Sinica, 2016, 56(2): 241-252.
[18] 聂 铭.不同连作年限地黄生长生理特性及其根区土壤化感物质研究[D].郑州:河南农业大学,2017.
[19] 田 美,刘汉湖,申 欣,等.百乐克(BIOLAK)活性污泥宏基因组的生物多样性及功能分析[J].环境科学,2015,36(5):1739-1748.
[20] 孙佳瑞,胡 栋,张翠绵,等.链霉菌S506对番茄苗生长和冷害生理生化的影响[J].中国农学通报,2012,28(31):167-171.
[21] 王世强.链霉菌JD211对水稻的防病促生效应及机制[D].南昌:江西农业大学,2014.
[22] 李欣欣,杨永庆,钟永嘉,等.豆科作物适应酸性土壤的养分高效根系遗传改良[J].华南农业大学学报,2019,40(5):186-194.
[23] 李芳柏,童 辉,陈鹏程,等.一株能转化重金属的贪铜菌属菌株及其应用[P].2016-03-16.
[24] 杨恩东,崔丹曦,汪维云.马赛菌属细菌研究进展[J].微生物学通报,2019,46(6):1537-1548.
[25] 康静静,杨玉荣,梁宏德.志贺氏菌病发病机制的研究进展[J].中国农业科学,2011,44(9):1939-1944.
[26] 杨 艳,王厚照,张 玲.多重耐药鲍曼不动杆菌氨基糖苷类修饰酶与16S rRNA甲基化酶基因的研究[J].微生物学杂志,2017,37(4):28-33.
