组蛋白甲基化修饰在多发性骨髓瘤发生发展中的作用研究进展*
2020-08-11
多发性骨髓瘤(multiple myeloma,MM)是以终末分化的浆细胞克隆性增生为特征的血液系统恶性肿瘤,可引起全身多脏器功能损害,主要表现为骨质破坏、肾损害、贫血、高钙血症及感染等。虽然MM的治疗取得较大进展,患者生存期获得延长,但易复发且不可治愈。本文旨在探讨MM的发病机制、寻找更为有效的治疗靶点。
MM的生物学特性错综复杂,是一种异质性很强的血液系统肿瘤,多种因素共同参与MM 的发生发展,主要包括遗传学异常[1]、骨髓微环境的改变[2-3]和表观遗传学异常[4]。近年研究显示,表观遗传学异常在MM 发生和疾病演变过程中扮演重要角色,包括DNA 甲基化、组蛋白修饰和非编码RNA。组蛋白修饰包括组蛋白乙酰化、甲基化、磷酸化、泛素化、类泛素化及瓜氨酸化。MM患者全基因测序结果显示,多种组蛋白甲基化酶发生突变,包括去甲基化酶UTX、甲基化酶MLL 和MMSET[5-6]。上述发现为MM 的联合化疗提供新的治疗靶点。组蛋白甲基化较为复杂,通常可发生在组蛋白N末端的赖氨酸(K)或精氨酸(R)残基上,其功能与位点(K 或R)和甲基化程度(单、二或三甲基化)相关,如H3K4me3、H3K36me3、H3K79me3、H4R3me1 和H4K20me1 可激活相关基因转录,而H3K9me3 和H3K27me3 则抑制基因转录[7-8]。组蛋白甲基化过程由组蛋白甲基化转移酶(histone methyltransferases,HMTs)和组蛋白去甲基化酶(histone demethylases,HDMs)共同调节,从而调控下游基因表达。现将组蛋白甲基化调节酶及其在MM中的作用综述如下。
1 HMTs
目前,已知的赖氨酸甲基化转移酶(lysine meth⁃yltransferases,KMTs)超过30 余种,分属KMT1~8 个家族(表1)。根据其结构特点分为两类,一类为含有SET结构域,另一类包含DOT1L结构域。精氨酸甲基化则主要由PRMTs(protein arginine methyltransfer⁃ase)家族成员催化完成,PRMTs 催化甲基基团从S-腺苷甲硫氨酸(SAM)转移到蛋白质精氨酸胍基的氮原子上。主要有3 个亚型:非对称精氨酸二甲基化(ω-NG,NG-asymmetric dimethylarginine,ADMA),对称精氨酸二甲基化(ω-NG,NG-symmetric dimethylar⁃ginine,SDAM)和精氨酸单甲基化(ω-NG-monomethy⁃larginine,MMA)。目前,已知有9 种PRTMs,分别为PRTM 1~9(表1)。
2 HDMs
组蛋白赖氨酸残基的去甲基化酶(histone lylsine demethylases,KDMs)分为两大类,一类为KDM1 家族成员,主要依靠FAD 为辅助因子完成去甲基化,如LSD1(lysinespecific histonedemethylase 1),以甲醛和非甲基化的赖氨酸残基为产物的去甲基化酶,可与co-REST、BHC80、HDAC1/2 等蛋白形成复合物发挥作用,作用于H3K4me1/2;另一类家族成员则包含1个JMJC(Jumonji C)结构域,依赖于Fe(Ⅱ)和α-酮戊二酸为辅因子,可对H3K4、H3K9和H3K36等多个位点进行去甲基化作用。包括多个亚型(KDM2~7)(表2)。目前,对精氨酸残基的去甲基化酶报道很少,有研究显示KDM 家族的部分成员,如KDM3A 和KDM6B,也可以在精氨酸残基去甲基化[9]。JMJD6(Jumonji domain-containing protein 6)为包含JMJC 结构域的铁及2-酮戊二酸依赖的加双氧酶,可对H3R2、H4R3 进行去甲基化。此外,组蛋白甲基化酶也会针对非组蛋白作用,从而影响重要的细胞信号通路,包括NF-κB、RAS 通路、PI3K/Akt、Wnt/βcatenin、P53和ERα通路[10]。

表1 HMTs及其作用位点
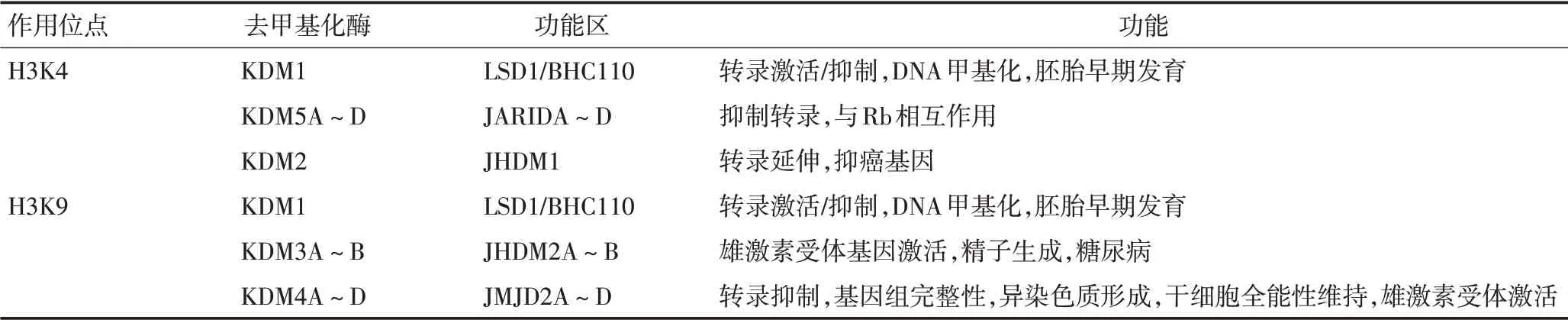
表2 KDMs及其作用位点

表2 KDMs及其作用位点(续表2)
3 组蛋白甲基化在MM发生发展中的作用
3.1 甲基化转移酶
MMSET 亦称为WHSC1/NSD2,是目前在MM 中研究最多的HMTs。约15%~20%MM 患者发生t(4;14)易位,导致IgH上MMSET和成纤维细胞生长因子受体3(FGFR3)过表达。t(4;14)易位患者无进展生存期(progression-free survival,PFS)和总生存率(overall survival,OS)显著缩短,但硼替佐米可克服这种不良预后[11]。MMSET主要催化H3K36双甲基化和H4K20 的双、三甲基化。MM 中MMSET 过表达导致H3K36me2 水平增高,引起癌基因转录激活,促进癌细胞增殖[12],受影响的基因还包括细胞周期(如cy⁃clin E2)、凋亡和P53通路(如BAX和Bcl2)、DNA修复(如ATM和GADD45A)和整合素介导的信号通路(如CDC42)[13]。既往研究报道MMSET还可以甲基化Au⁃rora激酶A(AURKA)导致P53蛋白酶体降解,从而引起肿瘤细胞增殖[14]。MMSET亦可与表观遗传学的抑制物相互作用,如sin3a、HDAC1-2和H3K4去甲基化酶LSD1/KDM1A,形成转录抑制复合物。MMSET、KAP1 和HDACs 形成的复合物可通过抑制miR-126导致c-myc 水平增高,刺激MM 细胞增殖[15]。沉默MMSET后可导致MM细胞周期停滞,诱导凋亡[16]。
MMSET 亦与耐药密切相关,使其成为一个潜在的治疗靶点。MMSET 可调节干扰素调节因子4(in⁃terferon regulatory factor 4,IRF4)的表达,抑制该基因可增强硼替佐米疗效。MMSET 高表达还使得DNA损伤修复增加,导致部分患者对DNA 损伤类药物不敏感。临床上也观察到表达t(4;14)患者在应用DNA损伤类药物后(如马法兰)很快复发[17]。MMSET在高效的同源重组和非同源末端连接两种DNA修复过程中是必需的,体外使其沉默后可增加对化疗药物的敏感性[18]。最近通过高通量筛选,5 种潜在的MMSET 抑制物被鉴定筛选,但仍需进一步工作确定其在MM中的作用[19]。
EZH2 为多疏抑制复合物2(polycomb repressive complex 2,PRC2)的催化亚基。其可催化H3K27me3,从而抑制靶基因转录,并参与调控细胞周期、细胞衰老、细胞分化等生理或病理过程[20]。近年EZH2成为MM中的研究热点,其在MM进展过程中上调,并与高危表型和不良预后相关[21]。EZH2过表达还与IL-6A、c-myc激活或miR26a下调相关。有研究通过全基因组测序详细阐释了MM中H3K27me3相关标志,发现了常见的激活(H3K4me3富集)和沉默(H3K27me3富集)的特殊靶基因,与疾病进展及不良预后相关[22]。应用EZH2特异性抑制剂UNC199和GSK343可重新激活参与分化、细胞周期和凋亡的相关基因起到抗MM作用。虽然有研究表明在MMSET高表达的MM细胞中EZH2抑制剂敏感性增强,但有研究[23]并未发现两者间存在关联,该结果尚需进一步研究。研究表明,UNC199还可通过影响miRNAs表达导致下游癌基因或活化因子下调[24]。EZH2也参与骨髓瘤骨病的发生发展。成骨前体细胞中转录抑制因子(growth factor independence 1,GFI1)介导的RUNX2沉默依赖于HDAC1、LSD1(KDM1A)和EZH2的募集[25]。这些研究成果提示其可能成为潜在的治疗靶点。多种EZH2抑制剂目前在实体肿瘤和淋巴瘤中已进入临床试验阶段,有单药应用亦有联合化疗方案。其中最具潜力的是tazemetostat/EPZ-6438,在淋巴瘤患者中观察到良好疗效,且不良反应较小[26]。而GSK2816126单药临床试验的结果欠佳,Ⅰ期临床试验入组复发的非霍奇金淋巴瘤、MM和实体肿瘤患者,起始剂量从50 mg开始,逐渐升至最大剂量3 000 mg。结果显示22例可评估的患者中,仅1例部分缓解和7例疾病稳定[27]。目前,已被中止试验。但相关生物标志物的鉴定可能会增加此类药物的临床疗效。Laurie等[28]发明一项基于基因表达的EZ评分体系,能够预测EZH抑制物在HMCL和初诊MM中的敏感性。
G9a和GLP属于Su(var)3-9家族,主要负责常染色质组蛋白H3K9位点的单、双及三甲基化进而抑制相关基因的转录。G9a 与胚胎发育、肿瘤细胞生长、认知及适应性行为和脂肪形成等多种生物过程密切相关[29]。近期研究显示,GLP在冒烟型骨髓瘤患者中高表达[30]。在急性淋巴细胞白血病、急性髓细胞白血病、淋巴瘤和MM 细胞系中,应用siRNA 技术或小分子抑制剂降低G9a表达可显著抑制瘤细胞生长,上述研究结果提示G9a/GLP 可成为MM 治疗潜在的新靶点。
KMT1/SUV39H1负责H3K9的三甲基化,在正常骨髓浆细胞和MM患者浆细胞中存在差异表达。H3K9甲基化是异染色质蛋白(heterochromatin protein1,HP1)染色区的停泊位点,在异染色质形成及基因转录调控中具有重要作用[31]。SUV39H1被发现可结合RUNX1的抑制区域。在MM中,高表达的SUV39H1与不良预后相关,敲除后导致增殖减低、凋亡增加、ROS过度产生和DNA损伤。SUV39H1抑制物chaetocin在人MM细胞系和原代细胞中具有抗瘤细胞作用[32]。
KTM2/MLL 可催化H3K4 甲基化,促进转录激活。MLL(mixed lineage leukemia)家族成员MLL1-5在7%MM 患者中发生突变,但对患者的PFS 和OS 均无显著性影响[33]。
PRMT5可催化组蛋白和非组蛋白精氨酸残基的甲基化[34]。PRMT5参与分化、增殖、同源重组和细胞迁移,在多种血液系统肿瘤和实体肿瘤中被发现过表达[35]。PRMT5被发现在MM中上调,并与不良预后相关。应用siRNA 敲低或抑制剂EPZ015666 可在HMCL 和原代细胞中抑制细胞周期,诱导凋亡。此外,口服的EPZ015666可在小鼠模型中有效降低瘤负荷。EPZ015666在淋巴瘤中可介导P53甲基化,但在MM中其甲基化并不依赖于P53[36]。相反,TRIM21被证实为PRMT5 在MM 中的结合物,导致经典NF-κB信号通路受抑,从而使IKK介导的自噬增强[37]。
PRMT4 作为精氨酸甲基转移酶1(coactivitor-as⁃sociated arginine methyltransferase1,CARM1)的辅助刺激因子,介导H3R2me2a、H3R17me2a、H3R26me2a和非组蛋白甲基化,从而促进转录。Drew 等[38]首次验证EZM2302,一种选择性PRMT4抑制剂,体外具有抗MM 作用。Nakayama 等[39]也于体外证明另一个PRMT4特异性抑制剂TP-064在MM中的抗增殖作用。
3.2 去甲基化酶
KDM1A也称为LSD1,介导H3K4me1/2去甲基化,抑制转录。但在特定条件下,KDM1A也可去除H3K9单双甲基,导致转录激活。KDM1A还可与其他表观遗传学调控复合物,如NuRD、SIRT1、CoREST/HDAC、MMSET和SIN3A/HDAC联合作用[40]。此外,KDM1A还可抑制P53功能[41]。KDM1A在多种血液系统肿瘤和实体肿瘤中表达增高[42],但KDM1A/LSD1在MM中的作用尚存争议。有研究发现症状型MM和浆细胞白血病(plasma cell leukemia,PCL)患者中LSD1的水平显著高于无进展MM患者,且E-钙黏附蛋白和波形纤维蛋白的水平降低;还可抑制破骨形成,增加MM细胞对HDAC抑制剂的敏感性;与MMSET形成抑制复合物,支持其在MM中的成瘤功能[43]。相反,Wei等[44]研究发现,KDM1A的种系突变会促进MM发展。此外,KDM1A在MM进展过程中下调,在MGUS 和MM 中KDM1A 也低于正常浆细胞;KDM1A突变的细胞中富含myc基因。KDM1A的抑制剂GSK-LSD1在小鼠模型中可促进MGUS发展;抑制KDM1A还可促进U266和原代细胞增殖。KDM1A/LSD1在MM中的作用如何仍需进一步研究。
KDM3/JMJD1C 赖氨酸去甲基化酶家族包括KDM3A、KDM3B 和JMJD1C。正常情况下,KDM3A和KDM3B 介导H3K9me1/2 去甲基化,在精子发育、干细胞自我更新和脂肪生成中发挥作用[45]。Ohguchi等[46]研究发现,KDM3A-KLF2-IRF4轴在MM 细胞存活中发挥重要作用。KDM3A 水平在MGUS 和MM 患者中较正常对照增高,敲除KDM3A 可起到抗MM 效应,并发现KLF2(Krüppel-like factor 2)和IRF4 下调。KLF2在支持正常的B细胞和浆细胞功能方面发挥作用。IRF4 为浆细胞特异性转录因子,可促进浆细胞成熟,且为MM 成瘤所必需[47]。有研究证实,KDM3A 在慢性缺氧条件下培养的MM 细胞中上调,敲除后可诱导细胞凋亡,进一步证实HIF1α 介导的KDM3A 上调可诱导长链非编码RNA MALAT1表达,进而上调糖酵解基因和抗凋亡途径,提示在MM进展过程中,MALAT 表达亦上调[48]。结果表明,HIF1α-KDM3A-MALAT1在缺氧的MM细胞中为潜在的治疗靶点。上述研究结束均表明,KDM3A 在MM 中有促成瘤的作用,因此需寻找特异性抑制剂[49]。
KDM5(JARID1)家族(KDM5A-D)介导H3K4me1-3去甲基,其中对H3K4me3亲和力最高,可直接抑制转录,或与其他抑制因子协同作用。3个独立的初诊MM患者队列的生存分析显示,KDM5B是预后不良的危险因素。全KDM5 抑制剂KDOAM-25 处理MM 细胞系MM1s,发现G1期停滞,细胞存活减少,ChIP-sequencing分析显示H3K4三甲基化水平增高[50]。另一种KDM5抑制剂compound33已被鉴定,但尚未应用在MM中[51]。
KDM6A(UTX)和KDM6B(JMJD3)介导H3K27me2/3去甲基化。UTX通常与其他表观遗传调控因子共同作用,如HMTs MLL2/3和HATs P300/CBP,激活转录[52]。已在10%MM患者中发现UTX表达的突变缺失[53]。在髓外MM和PCL,UTX的突变率达到30%~40%。Ohguchi等[54]研究发现,沉默UTX可促进瘤细胞增殖、黏附性和致瘤性,还可以增强MM细胞对EZH2抑制剂的敏感性,主要与Bcl-6重激活、继发IRF4和c-myc抑制有关。表明EZH2抑制剂治疗存在UTX突变的患者具有临床意义。而KDM6B/JMJD3则相反,其在MM中高表达,缺失后则可诱导细胞凋亡。NF-κB信号通路可激活KDM6A,依次上调MAPK通路相关基因,包括ELK和FOS,从而促进瘤细胞存活。KDM6B介导的MAPK通路活化是非去甲基化依赖的。KDM6B被鉴定为一种抑癌基因,在17p13缺失的高危患者中与TP53协同作用[55]。上述研究结果均证实KDM6A 可作为EZH2 抑制的标志,而KDM6B则可能成为潜在的MM治疗靶点。GSK-J4为首个KDM6家族的小分子抑制剂,但其抑制效果与KDM5类似[56]。因此,有必要开发特异性的KDM6抑制剂。
4 结语
组蛋白甲基化过程在MM中发挥重要作用,与表位突变、基因组不稳定、MM进展、耐药性出现和短暂的PFS 有关。深入研究组蛋白甲基化异常对MM 发病机制的影响,有助于寻找新的治疗干预靶点。针对HMTs 或HDMs 的药物已被证明可改变表观遗传格局,诱导抑癌基因重新表达。但临床应用的一项重要限制为不良反应。此外,表观遗传靶向药物对正常细胞群的长期影响亟需进一步研究。该类药物未来将进入个体化治疗,新的生物标志物识别、新药开发及多药联合等均可提供临床指导。
