用于肿瘤靶向治疗的糖基前药研究进展
2020-08-10谭云鹰傅俊杰尹健
谭云鹰,傅俊杰,尹健
(江南大学生物工程学院,江苏 无锡 214122)
传统化疗药物缺乏肿瘤细胞特异性,在杀伤肿瘤细胞的同时,对正常细胞的毒性也很大,易导致严重的系统毒副作用。此外,许多化疗药物存在水溶性差、生物利用度低等问题,限制了其临床应用。通过前药设计,将药物修饰为无毒的前体药物,经特定条件活化后释放出原药,是药物化学中常用的策略。抗肿瘤前药不仅可以改善药物的理化性质和药代动力学行为,更重要的是,通过合理的药物设计,可提高化疗药物的肿瘤靶向性,实现化疗药物的选择性、可控性释放,降低药物的毒副作用,提高其抗肿瘤活性。糖类作为生命基本物质之一,在前药设计中有着广泛的应用。糖基修饰不仅可以大大提高药物的水溶性,而且可以通过糖转运蛋白(GLUT)、糖苷酶等实现化疗药物的肿瘤细胞靶向性。本文对近年来糖基前药的研究进展作一综述,重点关注用于肿瘤靶向治疗的小分子糖基前药。
1 Warburg 效应
肿瘤靶向治疗的关键在于寻找肿瘤细胞与正常细胞的不同之处。20 世纪20年代,德国科学家Otto Warburg 发现了肿瘤细胞和正常细胞葡萄糖代谢方式的差异。正常细胞在氧气充足的条件下通过线粒体氧化磷酸化代谢葡萄糖并获取能量,在氧气供应不足时则利用无氧糖酵解产能。然而,肿瘤细胞即使在供氧充足的情况下,仍然以厌氧的糖酵解作为获取能量的主要方式。这一现象被称为Warburg 效应,Otto Warburg 因此获得1931年诺贝尔奖[1]。与氧化磷酸化途径(1 分子葡萄糖产生38分子ATP)相比,糖酵解(1 分子葡萄糖产生2 分子ATP)对葡萄糖的利用率大幅降低,而快速增殖、代谢旺盛的肿瘤细胞本身又需要大量的能量。因此,与正常细胞相比,肿瘤细胞对葡萄糖的依赖性大大增加,需要极大程度地提高葡萄糖摄取能力。研究表明,以GLUT 为代表的葡萄糖转运蛋白在多种肿瘤细胞表面高表达,以帮助肿瘤细胞摄取糖,这为肿瘤靶向提供了理论依据[2]。18F 同位素标记的2-脱氧-2-氟-D-葡萄糖(18F-FDG)因可选择性地在肿瘤部位聚集,自1980年起在临床上被用作造影剂诊断肿瘤,并沿用至今。受此启发,利用Warburg 效应靶向治疗肿瘤的策略也应运而生:将糖基与抗肿瘤药物偶联,得到的糖基修饰前药可被肿瘤细胞高表达的糖转运蛋白特异性识别,像“特洛伊木马”一样选择性地进入肿瘤细胞,经糖苷酶水解释放原药,实现靶向性抗肿瘤[3]。
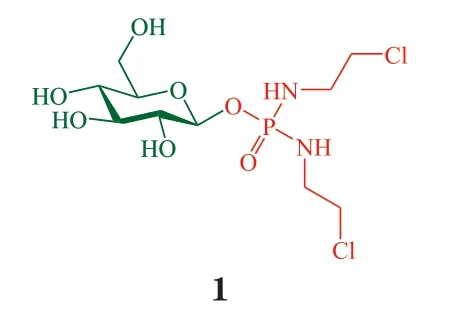
1995年Pohl 等[4]报道的葡磷酰胺(glufosfamide,1)是第1 个用于肿瘤靶向治疗的糖偶联药物。该药系将DNA 烷化剂异环磷酰胺(ifosfamide)的活性片段与D-葡萄糖以β-糖苷键偶联,利用GLUT进入肿瘤细胞,经β-葡萄糖苷酶水解后释放原药。MTT(溴化噻唑蓝四氮唑)实验中,GLUT 抑制剂根皮苷或根皮素(0.1 μmol · L-1)显著抑制葡磷酰胺对小鼠白血病L1210 细胞的增殖抑制活性,证明了前药依赖GLUT 进入肿瘤细胞。此后,多西他赛[5-6]、紫杉醇[7-8]、格尔德霉素[9]、酮洛芬[10]、苯丁酸氮芥[11-12]、去甲二氢愈创木酸[13]等相继被通过酯键、酰胺键、脲、丁二酸等连接臂与各种单糖偶联,得到各种糖基修饰的抗肿瘤药物[3,14]。
2 用于靶向性抗肿瘤的糖基前药
本节对近十年来基于Warburg 效应,利用GLUT 的小分子糖基前药设计作一介绍。此外,某些用于前药活化的糖苷酶也在肿瘤组织过表达,同样可被用于实现糖基前药的靶向性,也将在此节进行介绍。
2.1 利用糖转运蛋白的糖基前药设计
葡萄糖转运体包括促葡萄糖转运蛋白家族GLUT、钠依赖型葡萄糖转运体和最近发现 的SWEET(sugar will eventually be exported transporter),这3 种葡萄糖转运载体的生理功能和作用机制各异。迄今为止,对GLUT的研究最为深入。人源GLUT 由14 种亚型组成,其中11 种拥有催化葡萄糖顺浓度梯度运输的能力,这其中与肿瘤联系最为紧密的是GLUT1。人GLUT1 的晶体结构已于2014年被解析,其全长包含492 个氨基酸,共穿膜12 次,N 端和C 端都位于细胞质基质内。除了转运葡萄糖外,GLUT1 还能帮助细胞摄取半乳糖、甘露糖等,但正常情况下GLUT1 对其他糖类的亲和力小于葡萄糖。研究发现,GLUT1 表达量在转移性肝癌、肺癌、晚期胃癌、甲状腺癌等患者体内显著升高,这一特点已经被广泛用于糖基前药的设计[15]。糖基前药能够被GLUT 识别,经GLUT 介导进入细胞,从而实现肿瘤细胞靶向性。以下介绍近年来利用该策略设计药物的几个实例。
铂类药物因其作用机制明确、抗肿瘤谱广等优势,依然是如今抗肿瘤治疗的一线药物,代表药物顺铂(cisplatin)、卡铂(carboplatin)、奥沙利铂(oxaliplatin,OX)等已在全球各个国家获批上市,取得了良好的疗效。然而,由于铂类药物缺乏对肿瘤细胞的选择性,会破坏正常细胞的DNA,其严重的毒副作用(如肝肾毒性、胃肠道副作用、骨髓抑制等)长期以来限制了铂类药物的临床使用,成为该领域亟待解决的一大难题。此外,铂类药物水溶性极低,导致药物进入体内后不易排出,进一步增加了其毒副作用。利用在肿瘤细胞高表达的GLUT、特别是GLUT1,将其转运底物各种糖基与铂类药物的配体部分偶联,为提高铂类药物的肿瘤细胞靶向性、改善其水溶性、降低其毒副作用提供了有效策略。近年来,天津大学的高清志课题组在该方向的研究十分活跃。
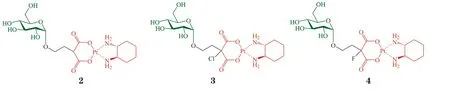
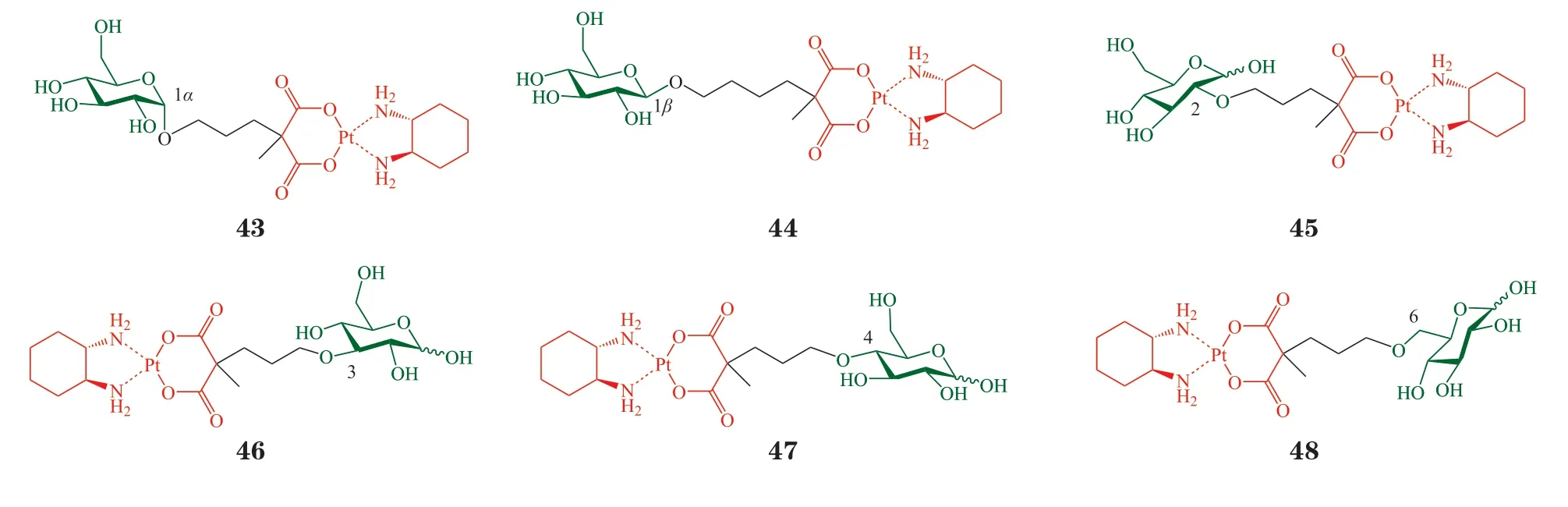
2013年,高清志课题组的Liu 等[16]基于奥沙利铂的结构,保留其螯合配体环己二胺,将其离去配体草酸用类似的丙二酸结构替换,进一步在丙二酸的α位引出2 个碳原子的连接臂,与葡萄糖形成α-糖苷键,得到了葡萄糖-铂偶联物(2)。丙二酸α位的另一个氢原子用氯或氟取代,分别得到衍生物3 和4。通过MTS 实验考察了化合物2~4 对6 株人肿瘤细胞(非小细胞肺癌A549 细胞、大细胞肺癌H460 细胞、卵巢癌SKOV3 细胞、乳腺癌MCF7细胞、结肠癌HT29 细胞、前列腺癌DU145 细胞)的增殖抑制活性。结果表明,上述3 种糖偶联物对6 株肿瘤细胞均表现出很高的毒性(IC50= 0.5~22 μmol · L-1),其中氟代产物4 的活性最佳,对HT29细胞的毒性(IC50= 0.45 μmol · L-1)至少是奥沙利铂的10 倍,对A549 和MCF7 细胞的毒性(IC50= 0.50 μmol · L-1)是奥沙利铂的2 倍。进一步用GLUT 抑制剂根皮苷考察糖转运蛋白对化合物4 活性的贡献。实验结果显示,根皮苷在高达1 mmol · L-1的浓度下,对HT29 和MCF7 细胞并无毒性,也不会与化合物4 相互作用,但是根皮苷可明显降低化合物4 的细胞毒性。例如,1 mmol · L-1根皮苷与化合物4 同时作用于HT29 细胞时,其IC50从单独给予化合物4时的0.45 μmol · L-1升高至约35 μmol · L-1;而相同条件下,根皮苷对奥沙利铂的细胞毒性影响很小。上述结果有力地证明了GLUT 对化合物4 抗肿瘤活性的重要作用,提示化合物4 通过GLUT 介导进入肿瘤细胞。研究人员进一步在动物实验中考察了该类化合物的安全性。BALB/c 裸鼠经尾静脉注射给药4次(分别于第1、7、14、21 d 给药)后测定最大耐受剂量(MTD)和半数致死剂量(LD50)。结果显示,化合物3 和4 的MTD(36 mg · kg-1)和LD50(约70 mg · kg-1)类似,且均约为奥沙利铂的5 倍。以LD50和HT29 细胞中的IC50的比值(LD50/IC50)作为治疗指数,化合物4 的LD50/IC50是奥沙利铂的30 多倍。上述结果充分表明,糖基修饰后因肿瘤细胞靶向性增加,铂类药物的安全性极大提高。此外,糖基的引入还大大提高了化合物的水溶性,化合物2~4 的水溶性分别是奥沙利铂的67、129 和155 倍。
亚 叶 酸(folinic acid,FOL)、5-氟 尿 嘧 啶(fluorouracil,F)和奥沙利铂三者联用(FOLFOX)是目前临床上肠癌的一线化疗方案。2015年,Li等[17]进一步考察了化合物4 在FOLFOX 化疗中的潜在应用价值。结果表明,亚叶酸或5-氟尿嘧啶均可与化合物4 发挥显著的协同抗肿瘤作用,效果与作用机制均与同奥沙利铂联用时一致。三组分联用时(铂类∶亚叶酸∶5-氟尿嘧啶 =1 ∶5∶10),含化合物4 的方案对5 株肿瘤细胞(H460、SKOV3、MCF7、HT29、DU145)效果比含奥沙利铂的方案更好。此外,在DBA/2 小鼠L1210 腹水瘤模型中,化合物4 的MTD 是奥沙利铂的6 倍,展现出更好的安全性,治疗效果(腹腔注射,每周1 次,3 周)比奥沙利铂更佳。该研究展现了糖偶联物4 良好的临床应用前景。
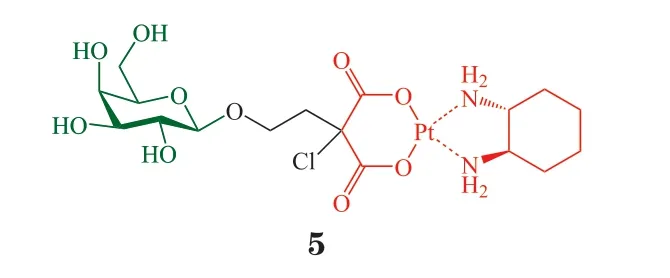
2016年,Wu 等[18]采用与Liu 等的方案类似的设计策略,将半乳糖与奥沙利铂通过丙二酸配体偶联,并在丙二酸α位引入氯原子,得到化合物5。MTS 实验表明,化合物5 对H460、HT29 的增殖抑制活性均优于奥沙利铂。对肺癌H460 细胞,化合物5 的活性(IC50= 10.04 μmol · L-1)比其葡萄糖类似物3(IC50= 22.10 μmol · L-1)更佳。GLUT 抑制剂槲皮素在无毒剂量下(20 μmol · L-1)显著减弱了化合物5(100 μmol · L-1)对HT29 细胞的毒性,细胞存活率由20%左右上升至50%左右,而对奥沙利铂的细胞毒性几乎无影响。该结果说明化合物5 的抗肿瘤活性依赖于GLUT。进一步研究发现,化合物5 可浓度依赖性地降低HT29 细胞对荧光底物2-脱氧-2-[(7-硝基-2,1,3-苯并二唑-4-基)氨基]-D-葡萄糖(2-NBDG)的摄取,效果与D-葡萄糖和2-脱氧-D-葡萄糖类似,而奥沙利铂对2-NBDG 的摄取无影响。该结果显示,化合物5 可与2-NBDG 竞争性地结合GLUT,证明化合物5 的肿瘤细胞转运依赖于GLUT 介导的糖基识别。体内抗肿瘤活性评价表明,在H460 肺癌移植瘤模型中,化合物5(iv,50 mg · kg-1,每周1 次,4 周)比顺铂(iv,3 mg · kg-1,每周1 次,4 周)效果更好;在HT29 结肠癌移植瘤模型中,化合物5(iv,50 mg · kg-1,每周1 次,4 周)比奥沙利铂(iv,7 mg · kg-1,每周1 次,4 周)效果更好。安全性评价表明,化合物5 的MTD 分别是顺铂和奥沙利铂的30 倍和14 倍。
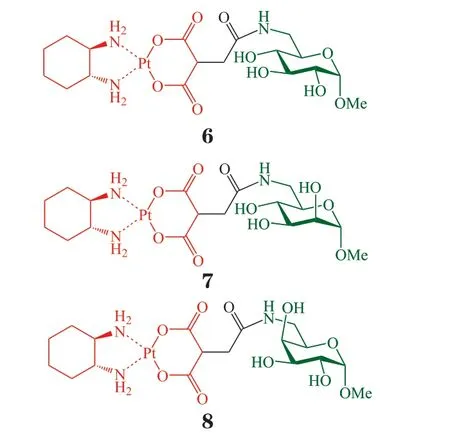
Li 等[19]在2016年又报道了6-氨基-6-脱氧-D-吡喃糖修饰的奥沙利铂衍生物。目标化合物6~8 的结构与前述化合物2~5 类似,分别含6-氨基-6-脱氧-D-葡萄糖甲苷、6-氨基-6-脱氧-D-半乳糖甲苷和6-氨基-6-脱氧-D-甘露糖甲苷结构。后续研究方法与上述例子类似,在此不再赘述。研究结果显示,6-氨基-6-脱氧-D-吡喃糖同样可以通过GLUT 介导的途径提高糖偶联物的肿瘤细胞靶向性、降低其毒副作用。
为了更清楚地评价GLUT1 对于糖-铂类偶联物细胞转运的贡献,Liu 等[20]在2017年通过shRNA转染构建了过表达GLUT1 的人肾脏上皮HEK-293FT 细胞和GLUT1 下调的HT29 细胞,用于评价甘露糖-铂偶联物9 的细胞毒性。结果表明,与正常的HT29 细胞相比,化合物9 对GLUT1 下调的HT29 细胞的毒性显著下降,而奥沙利铂对GLUT1表达水平不同的HT29 细胞的毒性相似。类似地,化合物9 对过表达GLUT1 的HEK-293FT 细胞的毒性显著高于对正常HEK-293FT 细胞的毒性,奥沙利铂未呈现该现象。这些实验结果表明,化合物9 的细胞毒性依赖于GLUT1。课题组进一步通过电感耦合等离子体质谱(ICP-MS)测定细胞内的铂含量,以评价不同细胞对于化合物9 的摄取量。结果显示,细胞与化合物9 孵育4.5 h 后,正常HT29 细胞内铂的含量是GLUT1 下调的HT29 细胞内铂含量的8 倍,证明化合物9 依赖于GLUT1 进入细胞发挥抗肿瘤作用。
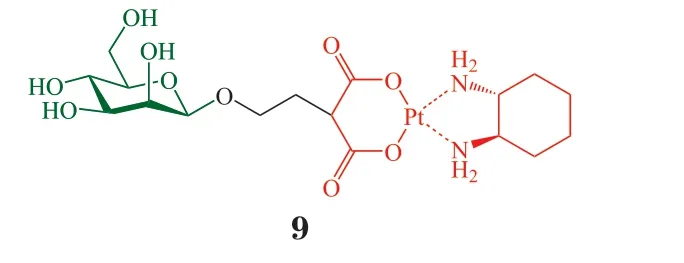
综上,高清志课题组近年来构建了各种糖-铂类偶联物,研究证实葡萄糖、半乳糖、甘露糖、6-氨基-6-脱氧吡喃糖等均可通过GLUT1 依赖的途径介导偶联物进入肿瘤细胞,实现靶向性抗肿瘤的目的。此外,糖基修饰提高了铂类药物的抗肿瘤活性、改善其水溶性、显著降低其毒副作用,为解决铂类抗肿瘤药物目前存在的问题提供了非常有效的策略。
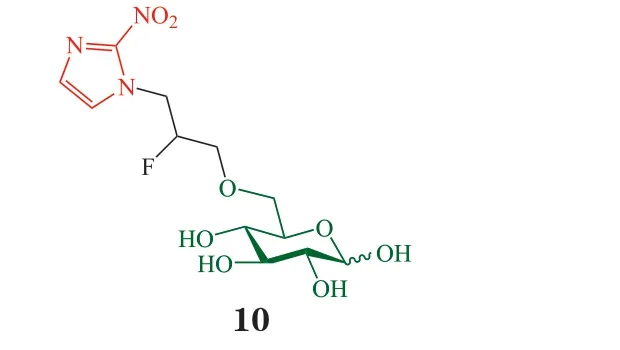
2-硝基咪唑,又称为氮霉素(azomycin),曾在临床试验中被用作放射增敏剂,但因毒性太大而失败。2012年,Kumar 等[21]合成了一系列2-硝基咪唑与葡萄糖6 位羟基的缀合物来克服该问题。该系列化合物在多种小鼠和人肿瘤细胞中均展现出低细胞毒性,IC50仅为毫摩尔浓度水平。该课题组在表达GLUT1 的非洲爪蟾卵母细胞中考察了上述化合物与14C 同位素标记的葡萄糖竞争被细胞摄取的能力。结果表明,含有三碳连接臂的化合物10 的摄取竞争能力最强,其至少是部分通过GLUT1 进入细胞。有意思的是,课题组在文中提到,低氧微环境可上调GLUT 的表达。鉴于许多肿瘤细胞处于低氧微环境,且低氧与肿瘤细胞的耐药密切相关,以GLUT 为靶点的糖类前药在低氧肿瘤治疗中的潜力令人期待。
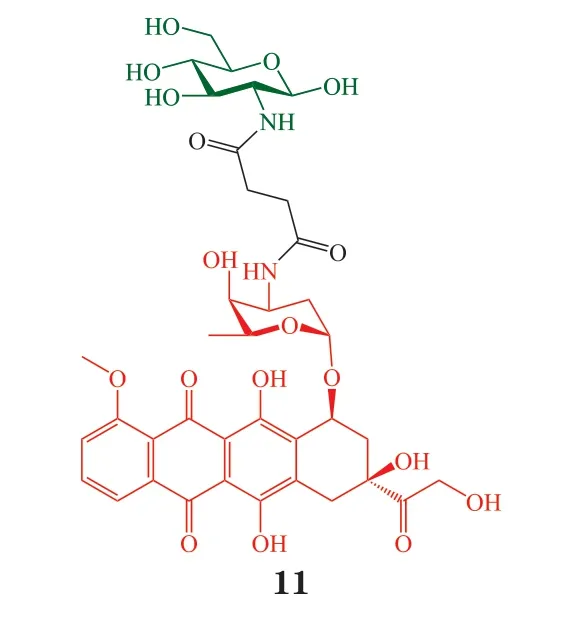
2013年,Cao 等[22]将阿霉素(DOX)与2-氨基-2-脱氧葡萄糖通过丁二酸相连得化合物11,以期利用糖基提高DOX 对肿瘤细胞的选择性,降低其毒副作用。对于肿瘤细胞,化合物11 与DOX 的活性相似;对正常细胞(人胚肺成纤维细胞),DOX 在低纳摩尔浓度下48 h 展现出显著毒性,而50 nmol · L-1的11 处理48 h 后未见细胞毒性。人肝癌HepG2 细胞经高浓度(25 或50 mmol · L-1)2-脱氧葡萄糖预处理后,对化合物11 的摄取显著降低,证明化合物11 至少部分通过GLUT 进入细胞。体内试验中,6 mg · kg-1的化合物11 与同剂量的DOX 对SKOV3移植瘤具有相似的抑瘤效果。
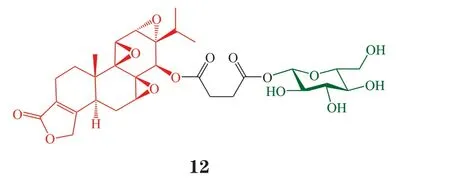
2016年,He 等[23]利用葡萄糖修饰雷公藤甲素(triptolide),为利用GLUT 设计糖偶联物提供了又一成功范例。雷公藤甲素是从中药雷公藤中分离得到的具有独特结构的二萜类化合物,被用于治疗炎症和自身免疫性疾病已有数百年历史。此外,雷公藤甲素在体外、体内实验中均表现出较强的抗肿瘤活性,最近研究表明,雷公藤甲素通过不可逆抑制转录因子IIH(TFIIH)的亚基XPB(xeroderma pigmentosum type B)发挥抗肿瘤活性。然而,雷公藤甲素及其衍生物的进一步成药开发在过去几十年中一直困难重重,主要障碍是系统毒性太大、水溶性太差。He 等合成了一系列雷公藤甲素的葡萄糖偶联物(glutriptolide),期望利用肿瘤细胞表面过表达的GLUT(特别是GLUT1 和GLUT3)实现化合物的肿瘤细胞靶向递送、降低其毒副作用。选择雷公藤甲素结构中最活泼的C14 位羟基,通过丁二酸酯、乙缩醛或直接连接的方式与葡萄糖形成β-糖苷键,共合成5 个化合物。经筛选发现,含丁二酸酯连接臂的前药12 在细胞外无XPB 抑制活性,提示糖基修饰可避免12 在被糖苷酶活化前发挥脱靶作用。细胞实验显示,化合物12 对HEK293T 细胞的增殖抑制作用最强,GLUT1 抑制剂WZB117 可拮抗化合物12 的增殖抑制活性,证明化合物12 的活性依赖于GLUT1。在2 株等基因肿瘤细胞系中进行的对比实验显示,化合物12 对过表达GLUT1 的人结直肠腺癌上皮细胞DLD1 的抑制作用强于野生型DLD1。与葡萄糖类似,化合物12 可浓度依赖性地抑制细胞对同位素标记的[3H]葡萄糖的摄取。这些实验均证实,GLUT1 可促进化合物12 的细胞摄取并实现肿瘤细胞靶向性。比较化合物12 对肿瘤细胞和正常细胞的抑制作用后发现,化合物12 对人呼吸道上皮细胞(AEC)、星形胶质细胞、人脐静脉内皮细胞(HUVEC)和人成纤维细胞的IC50在0.64~3.09 μmol · L-1之间,而对人前列腺癌细胞PC3、人肾癌细胞SK-RC-52、H460 和人非小细胞肺癌细胞H1975 的IC50在0.08~0.37 μmol · L-1之间。雷公藤甲素对上述正常细胞和肿瘤细胞的活性相似。该结果证实糖基修饰提高了化合物12 对肿瘤细胞的选择性。此外,得益于糖基的引入,化合物12的水溶性比雷公藤甲素大大提高。课题组给转移性前列腺癌模型小鼠腹腔注射化合物12,每日1 次,持续4 周后停药,结果显示雷公藤甲素组小鼠肿瘤立刻复发,而化合物12 给药组停药后2~3 周内仍未检测到肿瘤细胞,存活时间最长可达89 d。
除利用糖基和GLUT 的特异性识别外,利用糖基识别去唾液酸糖蛋白受体(ASGP-R)实现靶向性抗肿瘤也是常用的策略。然而文献调研发现,近年来,利用ASGP-R 的抗肿瘤研究绝大部分集中在纳米给药系统领域[24-25],不在本综述讨论的范畴内,故在此不作介绍。
2.2 利用糖苷酶的糖基前药设计
糖基前药需要在相应的糖苷酶的作用下水解释放出原药。研究指出,多种糖苷酶,如β-葡萄糖醛酸苷酶、β-半乳糖苷酶、α-甘露糖苷酶、β-N-乙酰氨基葡萄糖苷酶等,在不同肿瘤中表达升高,这为前药选择性地在肿瘤组织活化释放原药提供了理论依据,也为糖基前药的肿瘤靶向性提供了又一重保障。本节对近年来利用糖苷酶的小分子糖基前药设计进行介绍。
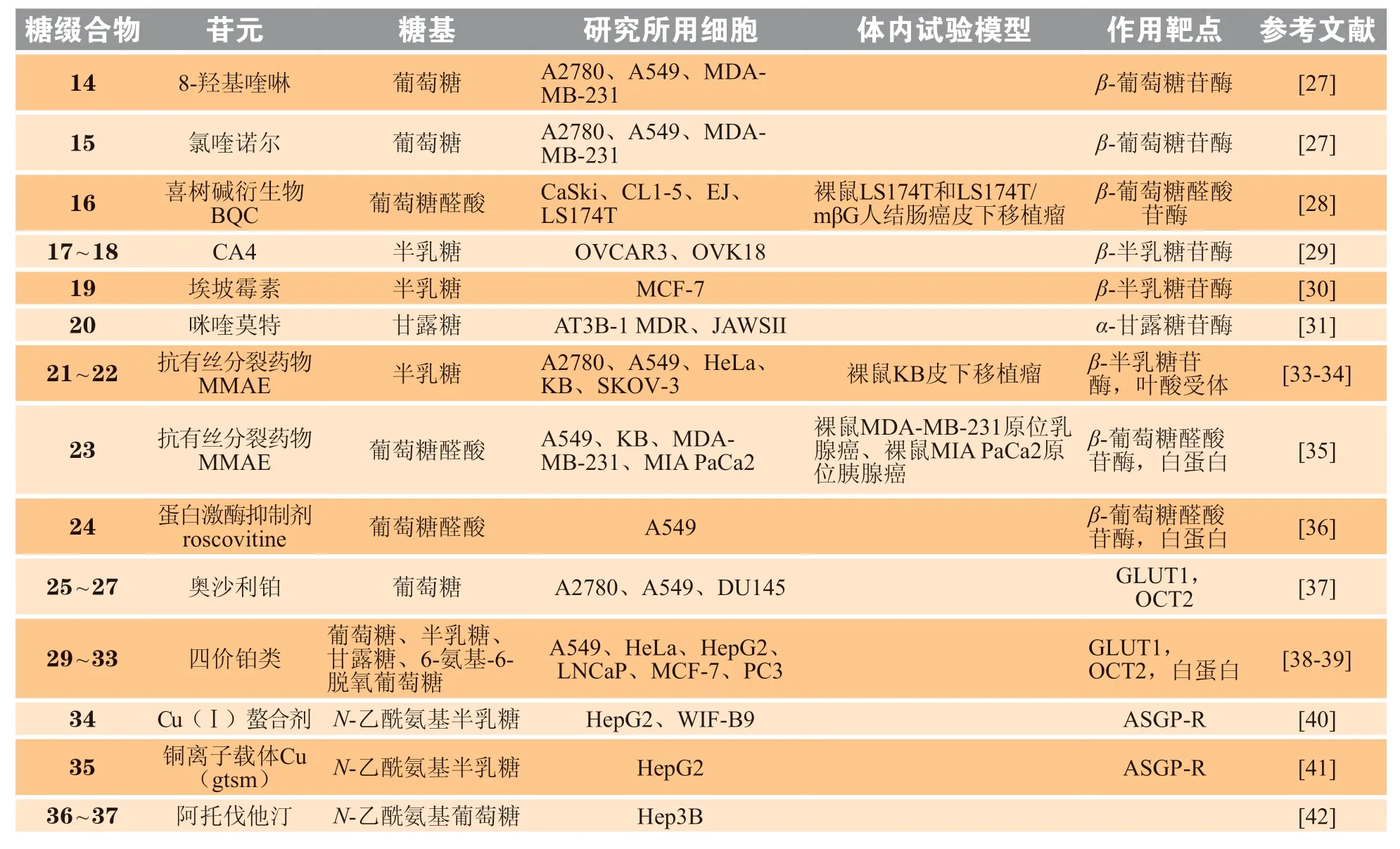
β-葡萄糖醛酸苷酶在多种肿瘤(如乳腺癌、肺癌、结肠癌、黑色素瘤等)区域含量升高。基于此,Papot 课题组在2011年报道了β-葡萄糖醛酸苷酶活化的前药设计。前药13 由葡萄糖醛酸、可自发降解的连接臂、DOX 和组蛋白去乙酰化酶(HDAC)抑制剂MS-275 组成。在β-葡萄糖醛酸苷酶作用下,β-葡萄糖醛酸苷键被切断,裸露出连接臂上的酚羟基,引发连接臂降解,通过1,4-消除和1,6-消除释放DOX 和MS-275。通过该策略,前药13 可在响应β-葡萄糖醛酸苷酶后释放2 种药物,发挥协同抗肿瘤活性。前药13 在pH 7 的磷酸缓冲液和牛血清中非常稳定,37℃下24 h 内未见降解。加入β-葡萄糖醛酸苷酶,前药按预想的机制迅速降解,在5 h 内释放出DOX,在18 h 内缓慢释放出MS-275。课题组用H290 肺间皮瘤细胞评价了前药13 的抗增殖活性,结果表明,前药13 对细胞无毒性,这可能是由于葡萄糖醛酸片段增加了整体分子的亲水性,使得前药无法进入H290 细胞。加入β-葡萄糖醛酸苷酶后,前药释放出DOX 和MS-275,可进入细胞发挥抗肿瘤作用,IC50达50 nmol · L-1,抗肿瘤活性远高于DOX(IC50= 372 nmol · L-1)、MS-275(IC50= 754 nmol · L-1) 以 及DOX 与MS-275 联 用(IC50= 221 nmol · L-1)的效果。在另外2 株人肺癌细胞H661 和H157 内也观测到了类似的现象。可见,前药13 并不是简单地释放出DOX 和MS-275,而是使两者发挥了更好的协同抗肿瘤作用。此外,1,6-消除释放的氮醌甲基化物活性中间体也对前药的抗肿瘤活性有着重要贡献[26]。
8-羟基喹啉(8-hydroxyquinoline,8-HQ)类化合物通过金属螯合(如二价铜离子)发挥诸如抗肿瘤和抗菌等多种生理活性,在有机和生物无机化学领域发挥重要作用。氯喹诺尔(clioquinol,CQ)是该类化合物中的代表,在20 世纪50年代即被广泛用于治疗腹泻和皮肤感染。但是在20 世纪70年代,出现了几千例由CQ 引起的亚急性脊髓视神经病变,导致CQ 被撤出市场,其毒副作用的产生可能是由于CQ 无选择性地将金属离子运输到中枢神经系统所致。经研究,CQ 还能够在微摩尔浓度下通过Caspase 依赖的凋亡途径发挥抗肿瘤作用,曾在Ⅰ期临床试验中被用于治疗复发性或难治的血液恶性肿瘤,但临床上如何避免非靶向的金属螯合仍是有待解决的问题。2012年Oliveri等[27]首次将8-HQ和CQ 的羟基与葡萄糖1 位连接,合成了2 个葡萄糖偶联物14 和15,期望通过葡萄糖提高药物的靶向性,避免非靶向的金属螯合带来的系统毒副作用。多种体外实验证明,糖偶联物14 与15 可被β-葡萄糖苷酶水解释放原药。细胞增殖抑制实验表明,与8-HQ 和CQ 相比,糖偶联物14 和15 直接作用于肿瘤细胞时抗肿瘤活性显著下降。额外加入铜离子可大大增加14 和15 的细胞毒性,最高可达无铜离子时的18.5 倍。
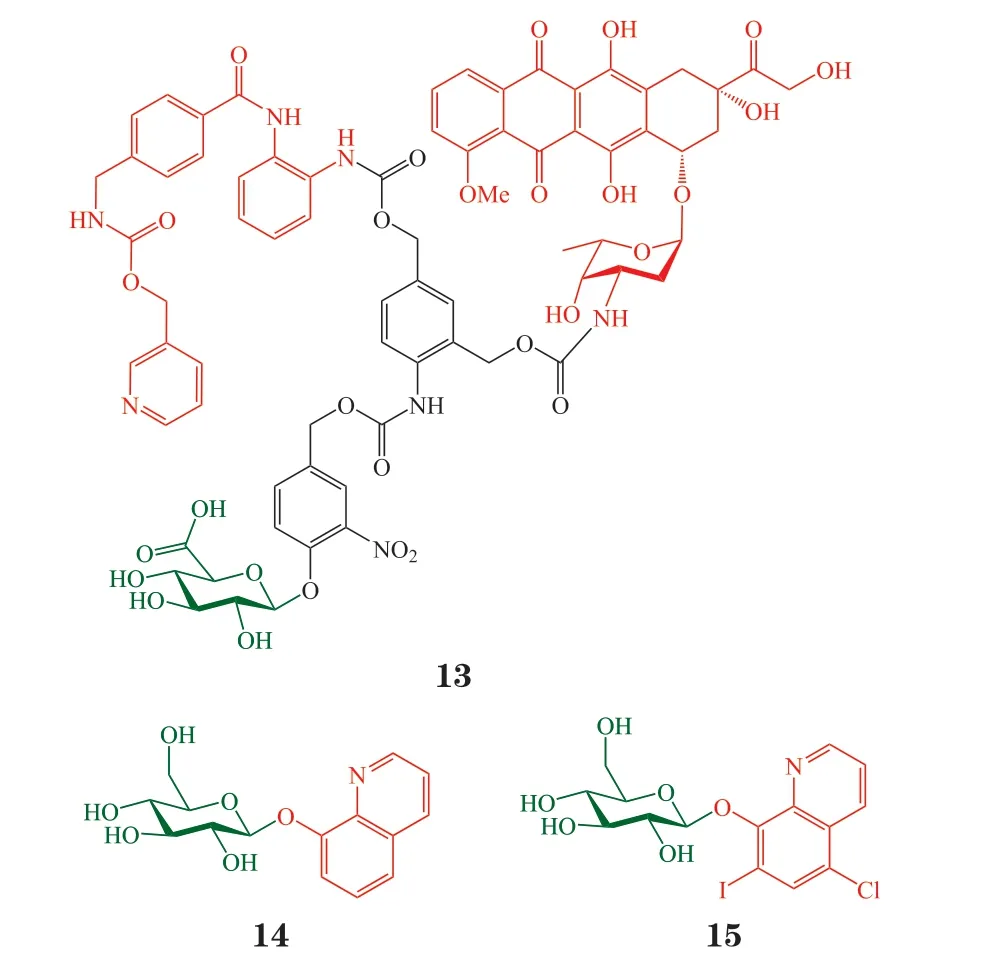
喜树碱(camptothecin)在纳摩尔浓度即可发挥抗肿瘤活性,极具临床开发前景,但其临床使用面临高毒性、低选择性、低水溶性等问题。此外,喜树碱可与人血清白蛋白(HSA)紧密结合,抗肿瘤活性被削弱。2016年,Prijovich 等[28]选择与HSA结合能力较弱的5,6-二氢-4H-苯并[de]喹啉喜树碱(BQC)为先导化合物,在其10 位羟基上通过可自发降解连接臂偶联葡萄糖醛酸,制备了前药16。与BQC 相比,糖偶联物16 在生理pH 下的水溶性由200 μg · L-1提 高 至775 mg · L-1,提 升 了 近4000倍。前药16 在磷酸盐缓冲液(PBS)和人血清中可稳定存在24 h 以上,加入低浓度的大肠埃希菌(E.coli)或人β-葡萄糖醛酸苷酶可催化前药16 快速水解释放BQC。课题组基于3H-胸腺嘧啶核苷掺入法考察了BQC 和前药16 的细胞毒性,结果表明,与BQC 相比,前药16 对多株人肿瘤细胞(肺腺癌细胞CL1-5、结直肠腺癌细胞LS174T、宫颈癌细胞CaSki、膀胱癌细胞EJ)的毒性显著下降(IC50为24.6~58.7 nmol · L-1)。加入1 μg 的β-葡萄糖醛酸苷酶后,前药16 的细胞毒性恢复至与BQC 相当的水平(IC50为1.3~1.8 nmol · L-1)。进一步考察HSA对前药16 细胞毒性的影响,结果发现,尽管HSA(40 g · L-1)的存在也会部分降低前药16 的细胞毒性,但在加入β-葡萄糖醛酸苷酶后,药物的IC50仍可达13.3 nmol · L-1,说明HSA 对前药16 活性的影响很小。课题组分别采用LS174T 和LS174T/mβG 人结肠癌移植瘤模型评估了前药16 的体内抗肿瘤效果。LS174T 模型中,细胞外的β-葡萄糖醛酸苷酶含量处于较低的自然状态水平,而LS174T/mβG 模型中,肿瘤细胞表面固定化了大量的β-葡萄糖醛酸苷酶。BQC(iv,10 mg · kg-1)和前药16(iv,25 mg ·kg-1)均可显著抑制肿瘤的生长,与对照组相比,给药后肿瘤体积显著变小,对小鼠体质量的影响很小。
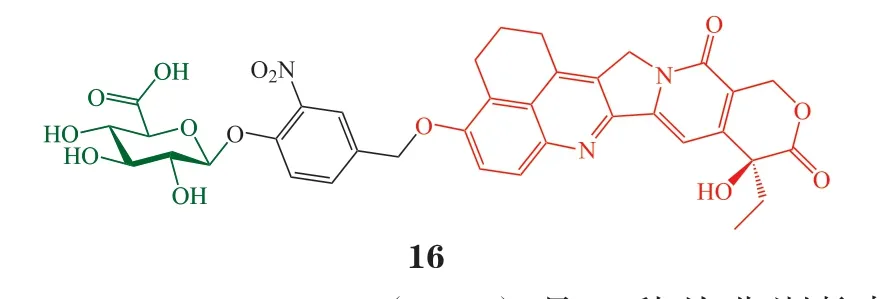
Combretastatin A4(CA4)是一种从非洲柳树树皮中分离得到的微管蛋白抑制剂,作用机制和紫杉醇类似,可用于治疗卵巢癌。β-半乳糖苷酶的活性在某些细胞株中显著升高,包括卵巢癌、乳腺癌、结肠癌、喉癌、神经胶质细胞瘤等。2017年,Suzuki 课题组将半乳糖与CA4 活性必需的B 环羟基直接、或通过连接臂偶联,得到半乳糖修饰的前药17 和18。体外微管蛋白抑制实验表明,前药17和18 在未经糖苷酶活化时无活性。HPLC 分析证实,加入β-半乳糖苷酶后,前药17 和18 均可释放CA4,且前药18 的释放速度更快。这可能是由于前药17 结构中β-半乳糖片段周围的空间位阻较大,不利于β-半乳糖苷酶接近,而前药18 结构中的连接臂减弱了位阻,使得糖苷酶更易与底物相互作用。课题组评价了前药17 和18 对卵巢癌细胞的增殖抑制活性。结果表明,前药18 对OVCAR3 和OVK18细胞的IC50分别为2.47和2.67 nmol · L-1,与CA4相当。然而,前药17 对2 株细胞的毒性显著降低,IC50分别为69.1 和252 nmol · L-1。前药17 和18 的体外细胞毒性差异可能源于前药与β-半乳糖苷酶的反应活性,也与前药的脂溶性和透膜能力相关。进一步研究发现,β-半乳糖苷酶抑制剂β-GA(1 mmol · L-1)可显著降低前药17 和18 的细胞毒性,证实前药对卵巢癌细胞的抑制作用依赖于β-半乳糖苷酶[29]。
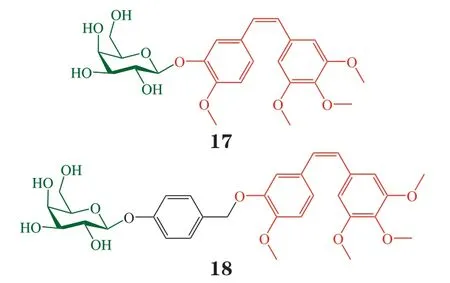
埃坡霉素(epothilone)是一种从微生物中发现的十六元大环内酯类化合物,与紫杉醇相同,具有促微管聚合活性,有望成为紫杉醇的理想替代品。然而已经报道的37 种天然埃坡霉素类化合物对正常细胞具有极高的毒性,对肿瘤的靶向性弱,且容易产生耐药性。2018年,Huang 课题组选用半乳糖,将其与埃坡霉素B 的7 号位手性羟基通过酯键相连,合成前药19。利用与抗体导向酶前药疗法(ADEPT)类似的策略评价前药的体外抗肿瘤活性。埃坡霉素B 和前 药19 对MCF-7 细 胞 的IC50分别为38.17 和5725.53 nmol · L-1,可见半乳糖基的偶联显著降低了药物的细胞毒性。加入曲妥珠单抗-β-半乳糖苷酶复合物后,前药被活化,释放出埃坡霉素B,活性提高约150倍,IC50恢复至与埃坡霉素B相同的水平[30]。
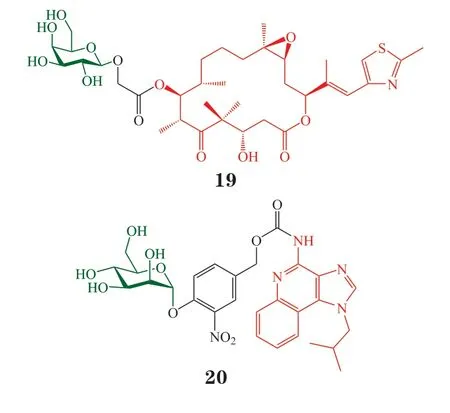
多药耐药(MDR)细胞可通过P-糖蛋白(Pgp)等将药物外排。因此,前药在肿瘤微环境活化释放出的活性原药可由目标细胞扩散至周围细胞,被称之为旁观者效应。旁观者效应固然有可能通过杀伤邻近的肿瘤细胞带来额外功效,但也可能带来难以预料的系统毒性。2018年,Mancini 课题组用免疫刺激剂代替传统化疗药物,构建了免疫刺激剂前药。前药活化释放出免疫刺激剂,并经P-gp 等介导外排后,通过旁观者效应活化邻近的免疫细胞产生免疫原性,而不产生毒副作用,达到了“扬长避短”的效果。选择咪喹莫特(imiquimod)作为免疫刺激剂,通过可自发降解的连接臂将咪喹莫特结构中喹啉上的氨基与甘露糖以α-糖苷键连接,得到前药20。之所以选择甘露糖修饰是因为文献报道α-甘露糖苷酶在多种肿瘤中高表达[31],可用于在肿瘤组织中选择性活化前药。选择同时高表达P-gp 和α-甘露糖苷酶的AT3B-1 MDR 前列腺癌细胞进行体外实验。LC-MS 分析发现,α-甘露糖苷酶(0.1 U ·mL-1)可在12 h 内将前药20 定量转化为咪喹莫特;AT3B-1 MDR 细胞可在48 h 内将60%的前药20 转化为咪喹莫特,半衰期为19 h。通过对咪喹莫特敏感的小鼠单核细胞白血病RAW-Blue 细胞证实,α-甘露糖苷酶与前药20 共孵育2 h 后可引发显著的免疫反应;AT3B-1 MDR 细胞与前药20 共孵育3 h 后可测得免疫反应。加入α-甘露糖苷酶抑制剂苦马豆素(100 μmol · L-1)可抑制免疫反应的发生,证实前药的活化依赖于α-甘露糖苷酶。最后,通过树突状细胞JAWSII 进一步验证前药活化后的免疫原性,结果表明,前药20 经AT3B-1 MDR 细胞活化后,可诱导JAWSII 细胞分泌白细胞介素(IL)-6、肿瘤坏死因子(TNF)、单核细胞趋化蛋白-1(MCP-1)等细胞因子,且分化度与直接用咪喹莫特诱导生成的细胞因子分化度类似[32]。
2.3 多功能多靶点的糖基前药设计
前文分别对利用GLUT 和糖苷酶设计糖基前药的研究进展进行了介绍。事实上,近年来,越来越多的研究并不是单单利用GLUT 或糖苷酶,往往是综合利用多种策略、设计多功能多靶点的前药分子,以进一步提高药物的肿瘤细胞靶向性、生物利用度和抗肿瘤活性。
多种肿瘤细胞表面高表达叶酸受体(folate receptor,FR),而绝大部分正常细胞表面无FR。Papot 课题组在2012年综合利用FR 和β-半乳糖苷酶,设计了第1 代无需靶向抗体的靶向抗肿瘤前药。前药21 围绕可自发降解的连接臂构建,同时包含半乳糖、叶酸和抗有丝分裂药物MMAE。前药21 经叶酸-叶酸受体介导的内吞选择性地进入肿瘤细胞,经溶酶体内β-半乳糖苷酶水解,裸露出连接臂上的酚羟基,诱发1,6-消除,最终释放出药物MMAE。肿瘤细胞和正常细胞的溶酶体中都存在β-半乳糖苷酶,特异性不足,但是通过靶向肿瘤细胞特异性的FR,提高了前药的肿瘤靶向性。体外细胞实验显示,前药21 的细胞毒性与细胞表面的FR 水平密切相关。对于高表达FR 的口腔上皮癌KB 细胞,前药21 与MMAE 的IC50一致(0.240 nmol · L-1);对于FR 水平略低的宫颈癌HeLa 细胞,前药21 的活性(IC50= 8.408 nmol · L-1)低于MMAE(IC50= 0.630 nmol · L-1);对于不表达FR 的A549 细胞,前药21的活性(IC50= 195.230 nmol · L-1)远低于MMAE(IC50=0.872 nmol · L-1)。以上结果表明前药21 可选择性进入高FR 表达的细胞。进一步实验表明,前药21可抑制Hela 细胞的微管蛋白聚集,效果与MMAE一致,证明前药21 可在HeLa 细胞内经β-半乳糖苷酶水解释放原药。此外,前药21 在表达FR 的细胞内释放MMAE 后,可通过旁观者效应杀伤周围的不表达FR 的肿瘤细胞。课题组用荧光素酶转染的裸鼠模型评价了前药21的体内抗肿瘤活性,结果表明,前药21(iv,5 mg · kg-1)治疗组从第21 d 开始荧光彻底消失,代表肿瘤完全消失,效果优于MMAE。MMAE 高剂量组可引起小鼠体质量下降,治疗结束后7 只小鼠仅存活3 只,而前药21 治疗组未见明显毒性,31 d 后7 只小鼠全部存活。体内实验证明前药21 的安全性和有效性相比于MMAE 均得到了提高[33]。
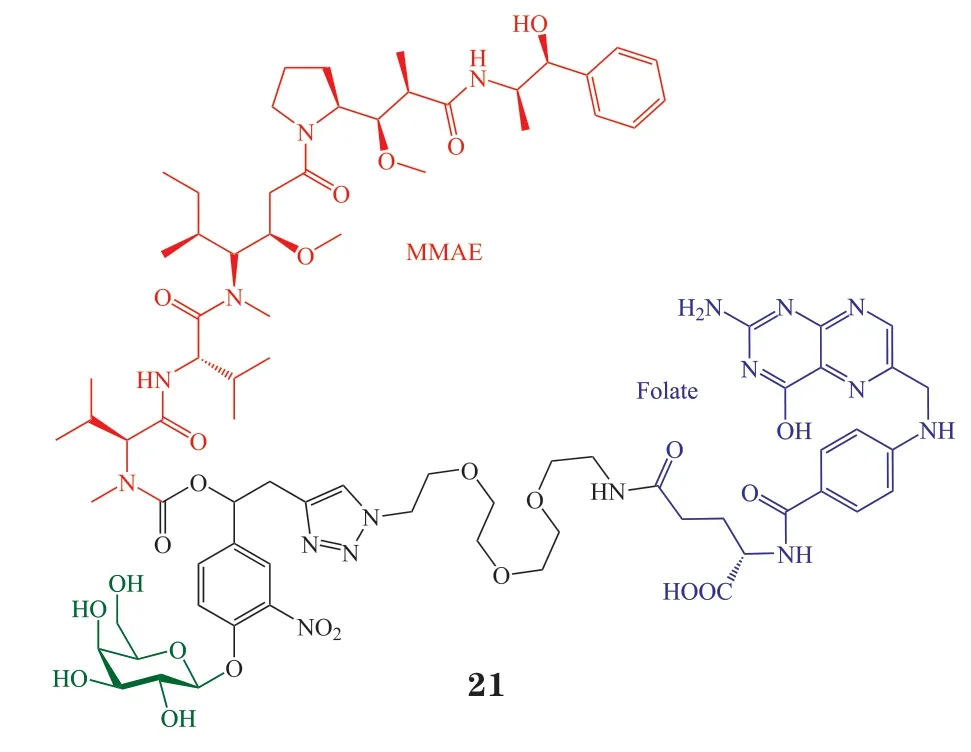
2015年,Papot 课题组又对前药21 的结构进行了进一步改造,得到前药22。前药22 经β-半乳糖苷酶水解后,可通过1,6-消除和1,4-消除释放2 分子MMAE,发挥更强的抗肿瘤活性。用HPLC 和高分辨质谱(HRMS)监测了前药22 在β-半乳糖苷酶作用下(pH 7.2,37 ℃)的降解情况,结果显示,前药22 可在2 h 内完全释放MMAE。细胞实验测得,前药22 对FR 表达水平较低的HeLa、SKOV3 和人卵巢癌细胞A2780 依然有很强的毒性(IC50为9.62 ~64.51 nmol · L-1),优于前药21(IC50为20.16~248.23 nmol · L-1)。进一步研究表明,前药22 在A2780 细胞内释放的MMAE 是前药21 释放量的5 倍[34]。
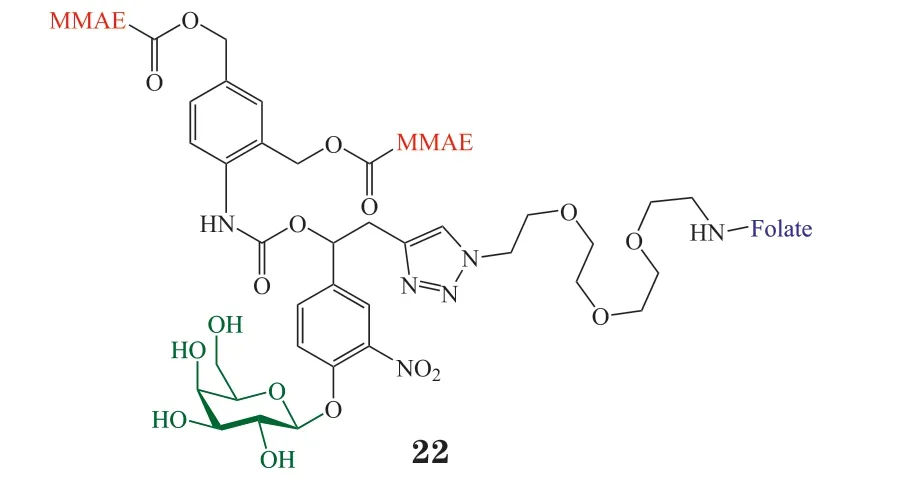
2017年,该课题组又报道了前药23,其结构与前药21 类似,区别在于前药23 用含有马来酰亚胺的侧链替代了前药21 结构中的叶酸,用葡萄糖醛酸替代了前药21 中的半乳糖。进入血液循环后,马来酰亚胺片段可通过Michael 加成与白蛋白34 位半胱氨酸结合。与白蛋白结合避免了前药的快速肾清除,且由于肿瘤血管特有的病理特征,结合物可通过被动积累选择性地富集于肿瘤组织,经β-葡萄糖醛酸苷酶水解后释放MMAE。该策略无需利用肿瘤细胞表面的特异性受体(如FR)即可实现靶向性。细胞实验表明,前药23 对KB、A549、乳腺癌细胞MDA-MB-231 和胰腺癌细胞MIA PaCa2 的毒性显著低于MMAE,加入β-葡萄糖醛酸苷酶后毒性恢复至与MMAE 同一水平。安全性评价发现,BALB/c小鼠对前药23 的耐受剂量高达8 mg · kg-1,远高于MMAE 的0.75 mg · kg-1。用MDA-MB-231 原位乳腺癌模型评估了前药23的体内抗肿瘤活性。结果显示,前药23(iv,4 mg · kg-1,每周1 次,5 周)显著降低了肿瘤的体积,效果优于MMAE(iv,0.5 mg ·kg-1,每周1 次,5 周)治疗组,且不引起体质量降低等毒副作用。进一步用MIA PaCa2 原位胰腺癌模型考察前药23 的体内活性,结果显示前药23 的治疗效果仍然优于MMAE,且对于晚期、伴随严重肿瘤乏氧的胰腺癌原位瘤仍有很好的治疗效果[35]。
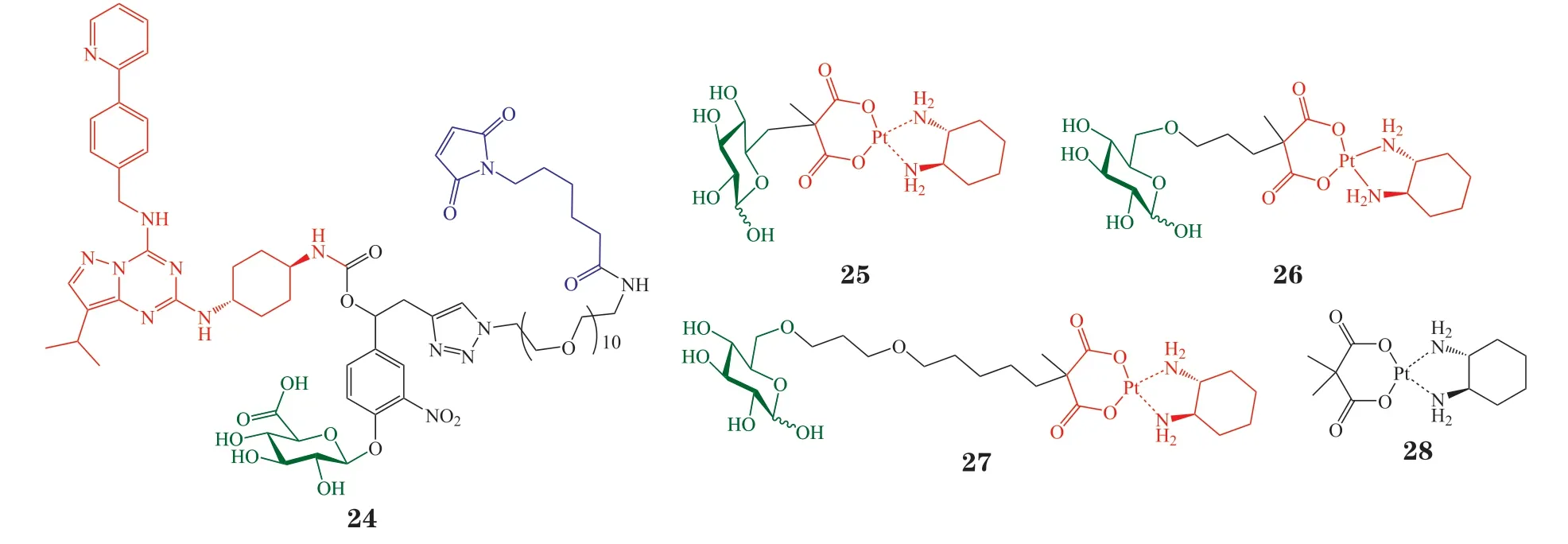
2018年,该课题组利用相同的策略,合成了蛋白激酶抑制剂roscovitine 衍生物的葡萄糖醛酸前药24,以期降低蛋白激酶抑制剂普遍存在的严重毒副作用。实验结果指出,前药24 在200 nmol · L-1的浓度下对A549 细胞无毒性,而原药的IC50为10.3 nmol · L-1,加入β-葡萄糖醛酸苷酶后,前药24 的细胞毒性和激酶抑制活性恢复[36]。
前已述及,高清志课题组利用Warburg 效应,开发了一系列糖-铂类偶联物。此外,近年来其他课题组围绕糖基修饰的铂类药物也有一些独特的发现。2016年,Lippard 课题组合成了3 个葡萄糖-铂偶联物。从铂离去配体结构中丙二酸的α位引出长度不同的连接臂,与葡萄糖6 位偶联,得前药25~27。之所以选择与葡萄糖6 位连接是因为,对与GLUT1同源的木糖转运蛋白XylE 的分析发现,D-葡萄糖的6 位不参与糖和转运蛋白间的氢键作用,因此,C6 修饰不会干扰糖偶联物与GLUT1 的结合。细胞摄取研究发现,肿瘤细胞A2780、DU145 和A549对前药25的摄取最为有效,随着连接臂长度的增加,摄取量降低,这可能是由于空间上太长的结构阻碍了底物与GLUT1 结合后进入细胞所必需的构型反转。苷元28 的脂溶性远大于前药25,理应更容易透膜,但与细胞作用8 h 后,前药25 在细胞内的含量显著高于苷元28,充分说明了糖基对于细胞摄取的贡献。选用高表达GLUT1 的DU145 和肾癌A498细胞,以及相对应的正常前列腺细胞RWPE2 和肾上皮细胞CCD1105,以进一步比较肿瘤细胞和正常细胞对前药25 摄取的区别。结果表明,DU145 对于前药25 的摄取量是RWPE2 细胞的5 倍。以上结果均提示GLUT1 在前药转运中的重要作用。然而作者并未止步于此,而是进一步考察了其他有可能介导前药转运的靶点。有机阳离子转运体2(OCT2)在某些肿瘤细胞内过表达,且在含有反式环己二胺结构的铂类化合物的细胞摄取中发挥重要作用。研究发现,A2780 细胞中,GLUT1 抑制剂4,6-O-乙缩醛-α-D-吡喃葡萄糖(EDG)将前药25 的细胞摄取降低50%,OCT2 抑制剂西咪替丁(Ctd)导致前药25 的细胞摄取降低45%,EDG 和Cdt 同时作用共降低65%。该结果提示,除GLUT1 外,OCT2也在前药25 的细胞摄取中发挥重要作用,这一结论是其他糖-铂类偶联物研究中没有考虑到的。在GLUT1 和OCT2 的协同作用下,前药25 展现了优异的肿瘤细胞选择毒性,对高表达GLUT1 的神经细胞Neuro-2A 的毒性比对肿瘤细胞低得多[37]。
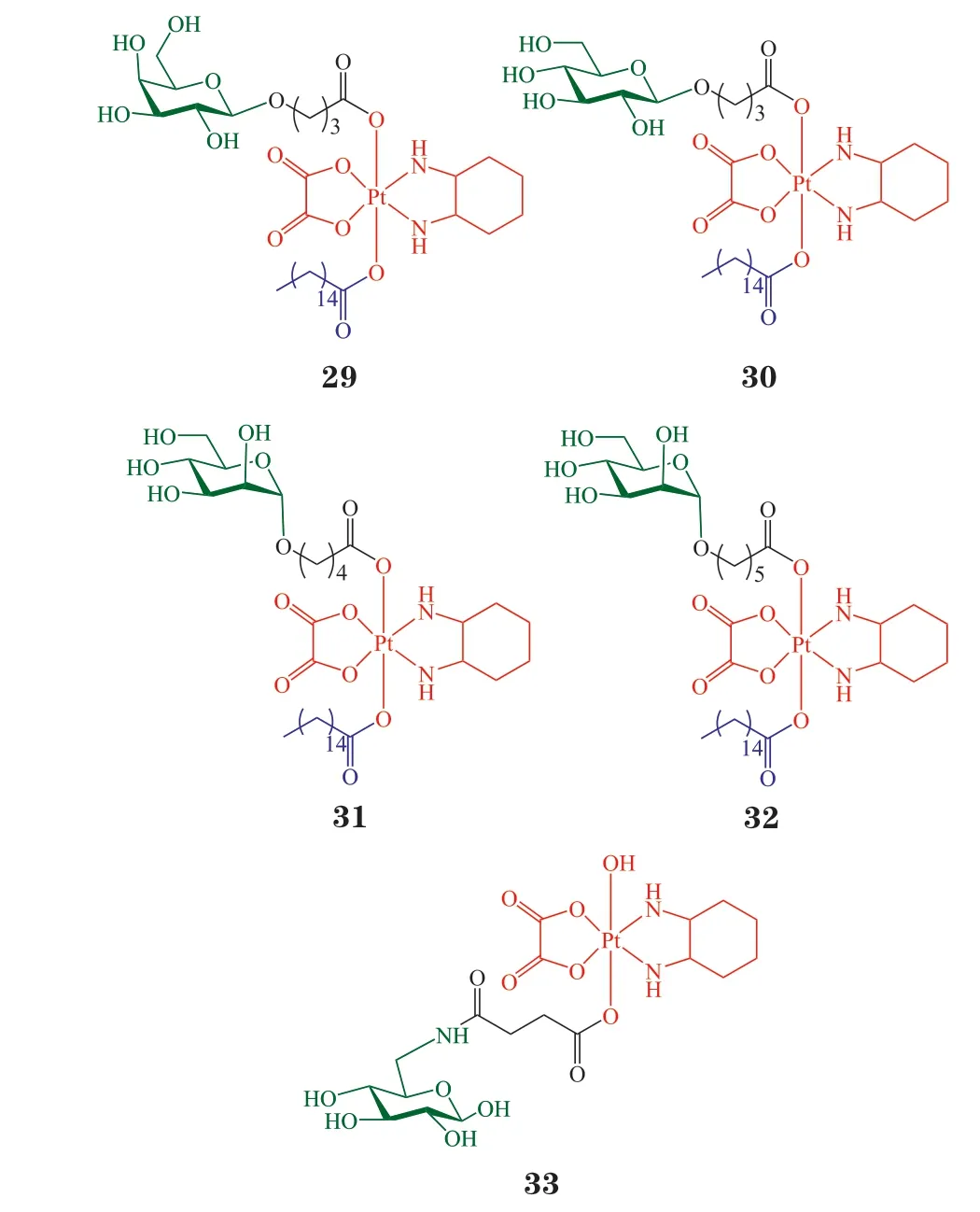
前述铂类药物都是二价铂结构,易于被血液中的亲核试剂进攻破坏,影响其生物利用度。王鹏课题组率先研究了四价铂类糖基偶联物。低自旋八面体结构的Pt(Ⅳ)复合物在血液中不易水解或发生配体取代,稳定性高于Pt(Ⅱ)。此外Pt(Ⅳ)可被肿瘤细胞内高浓度还原物质如抗坏血酸、谷胱甘肽等还原,释放Pt(Ⅱ),增加了药物的肿瘤细胞选择性。2017年,课题组的Ma 等[38]设计合成了一系列糖-Pt(Ⅳ)偶联物,以期同时利用GLUT和OCT2 靶向肿瘤细胞。此外,在结构中引入可与HSA 作用的十六碳链,进一步提高前药的血浆稳定性和肿瘤靶向性。MTT 筛选发现,糖基C1 位偶联的化合物29~32 的抗肿瘤活性优于C6 偶联的化合物,对HeLa、MCF7、前列腺癌LNCaP、PC3、肝癌HepG2、A549 等肿瘤细胞的毒性(IC50为0.24~3.97 nmol · L-1)是阳性对照药顺铂、奥沙利铂和赛特铂(satraplatin)的100 多倍,且对耐药细胞A549R 也有很好的增殖抑制效果(IC50为0.65~3.52 nmol · L-1)。GLUT1 抑制剂EDG 和根皮苷可显著降低化合物30的肿瘤细胞毒性和细胞摄取,而对C6 偶联物的影响很小,说明C6 偶联物几乎不依赖于GLUT1,这与上述Lippard 课题组的结论相悖,可能是因为Pt(Ⅳ)(八面体)与Pt(Ⅱ)(正方形平面)的空间结构不同,因此偶联物上的糖基与GLUT1 结合的位置不同。OCT2 抑制剂Ctd 显著影响化合物30 的细胞毒性和摄取,不影响化合物29、31 和32,说明只有化合物30 可同时作用于GLUT1 和OCT2。更为重要的是,研究证实化合物30 可被犬肾上皮细胞MDCK 上的GLUT1 和OCT2 受体识别。MDCK 细胞常用于评价药物是否适合口服给药。因而,该结果提示化合物30 具有开发为口服抗肿瘤药物的潜力。进一步研究表明,化合物29~32 对正常细胞3T3 和293T 的毒性明显弱于对肿瘤细胞HeLa 和MCF-7 的毒性,具有良好的肿瘤细胞选择性。MCF-7 体内模型实验显示,化合物30 在肿瘤组织中的积聚量分别是化合物29、32 和赛特铂的7.2、8.9 和3.2 倍。体内抑瘤实验发现,化合物30(iv,5 mg · kg-1)可有效延缓肿瘤生长,效果与奥沙利铂相当,但毒性低于奥沙利铂。2018年,该课题组又报道了能同时作用于GLUT1 和OCT2 的糖-Pt(Ⅳ)偶联物33[39],其研究方法及结果与上述研究类似,在此不再赘述。
2.4 用于其他疾病靶向递药的糖基前药设计
如前所述,糖基前药主要用于实现肿瘤的靶向性治疗,但也不局限于此。糖基受体种类繁多、功能多样,药物通过糖基修饰,利用糖基和受体之间的特异性识别也可以在其他疾病中实现药物的靶向递送。这其中应用最为广泛的是在肝细胞表面特异性表达的ASGP-R。ASGP-R 可识别半乳糖、乳糖、N-乙酰氨基半乳糖(GalNAc)等,对GalNAc 的亲和力最强[解离常数(Kd)= 10-5L · mol-1]。因此,药物经GalNAc 修饰可实现肝靶向递送,用于选择性治疗多种疾病。
人体内的铜离子受到精密调控,以保证维持酶功能所需浓度而又不因铜离子浓度过高而产生毒性。然而,威尔逊病患者先天性铜代谢障碍,缺乏将铜离子排出肝细胞的能力,导致肝细胞内铜过载而威胁生命,需要长期接受铜螯合剂治疗。传统铜螯合剂存在毒副作用大、有效性不高等问题。2012年,Delangle 课题组以基于次氮基三乙酸结构的三足配体为Cu(Ⅰ)螯合剂,通过二硫键与Gal-NAc 偶联,设计合成了前药34。利用肝细胞表面ASGP-R 对GalNAc 的特异性识别,前药34 可被靶向运输至肝细胞,在细胞内还原性物质作用下断开二硫键释放配体螯合铜离子。实验表明,前药34 本身无Cu(Ⅰ)螯合能力,经谷胱甘肽作用后可螯合Cu(Ⅰ)。利用荧光标记的34 类似物发现,前药可选择性进入表达ASGP-R 的HepG2 和大鼠肝癌/人成纤维细胞杂交细胞WIF-B9,不进入不表达ASGP-R 的HeLa细胞,证明了GalNAc 的肝靶向作用。用免疫荧光法监测发现,前药34(10 μmol · L-1)作用3 h 后可逆转WIF-B9 细胞内的铜过载[40]。
铜离子缺乏时,也会引起一系列疾病,如非酒精性脂肪肝等,解决办法是利用铜离子载体补充铜。然而,传统的铜离子载体缺乏组织特异性,容易造成正常组织中的铜离子聚集,引发氧化应激和损伤。2018年,Chang 课题组利用GalNAc 作为肝靶向配体,与铜离子载体Cu(gtsm)通过β-糖苷键连接,合成了前药35。细胞实验结合ICP-MS 分析表明,前药35 可选择性地将铜离子带入高表达ASGP-R 的HepG2 细胞,而不影响无ASGP-R 表达的HEK-293T细胞。温度由4℃升高至37℃时,HepG2 细胞对前药35 的摄取显著增加,说明前药35 通过ASGP-R介导的主动运输进入细胞。D-半乳糖(1 mol · L-1)竞争性抑制前药35 的铜递送,进一步证实了ASGP-R依赖的作用机制。以表达荧光素酶的转基因小鼠结合铜离子响应的荧光素探针考察前药35 的体内靶向递送能力,结果证明前药35 可显著增加肝内的铜离子浓度。组织分布实验显示前药35 能使得肝脏内铜离子含量6 h 后提升300%,且不影响其他器官中的基础铜离子含量,而无糖基修饰的Cu(gtsm)脱靶作用明显,显著影响肾、心脏、肺和脑中的铜离子含量[41]。
除 上 述2 例 利 用GalNAc 靶 向ASGP-R 外,有报道指出N-乙酰氨基葡萄糖(GlcNAc)可通过钙离子依赖的模式与肝细胞结合。基于此,2017年,Dong 课题组制备了肝靶向的阿托伐他汀(atrovastatin)前药。阿托伐他汀通过抑制肝细胞内的3-羟基-3-甲基戊二酰辅酶A(HMG-CoA)还原酶发挥调血脂作用,但水溶性差、透膜能力弱,因而生物利用度很低。通过点击化学将GlcNAc 与阿托伐他汀偶联,制备了前药36 和37,以期提高其肝细胞靶向性。连接GlcNAc 后,前药36 和37的水溶性较阿托伐他汀分别提高了4 倍和9 倍。细胞摄取实验表明,人肝癌细胞Hep3B 对36 和37的摄取量分别是阿托伐他汀的1.76 倍和2.07 倍,且质谱分析证实前药36 和37 能够在细胞内释放出阿托伐他汀。课题组测定了前药36 和37 在细胞内活化后抑制HMG-CoA 进而诱导低密度脂蛋白表达的能力,结果显示两者均能诱导Hep3B 细胞内的低密度脂蛋白表达[42]。
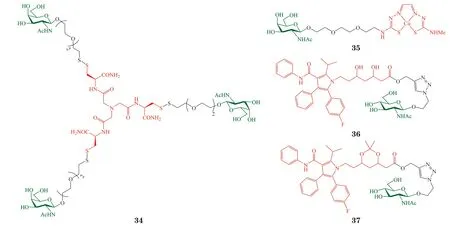
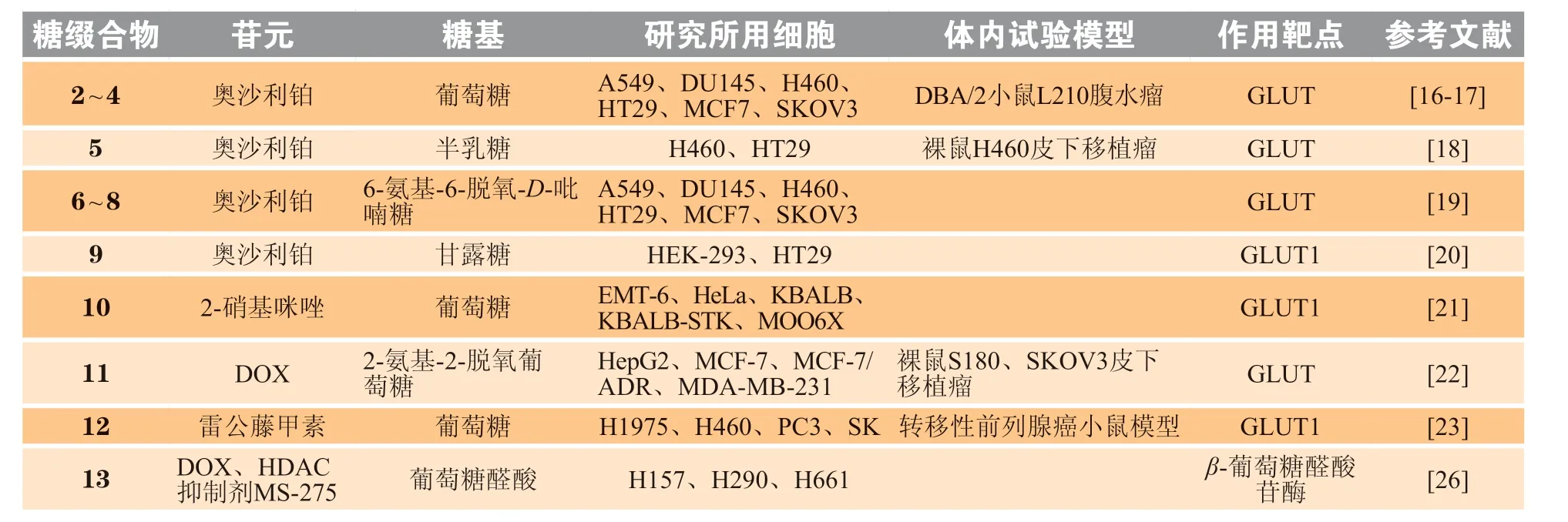
表1 用于肿瘤靶向治疗的糖基前药一览Table 1 Glycoconjugate prodrugs for targeted cancer therapy
表1 概括了各靶向性糖基前药的关键信息。
续表1
3 展望
糖基前药在靶向性抗肿瘤领域已经展现出令人期待的前景。然而,为了更好地发挥其价值、推动临床转化,在今后的研究中还有若干问题值得关注。
3.1 如何准确评价糖基对于前药靶向性的贡献
如“2.1”节所述,糖基前药可经GLUT 介导的转运进入细胞,实现药物的肿瘤细胞靶向性,这是许多糖基前药设计的理论依据。值得注意的是,糖基修饰往往还有多重作用,如提高药物的水溶性、改善其药代动力学行为等。此外,糖修饰位点的不同、苷元结构和性质的不同等都会直接影响糖偶联物与GLUT 的结合。因此,一个不容忽视且非常关键的问题是,糖基前药到底是不是经GLUT 进入肿瘤细胞?GLUT 对前药的肿瘤靶向性到底有多少贡献?“2.3”节中Lippard 课题组这一问题的回答做出了引领性的示范[37]。前药25 事实上通过GLUT1和OCT2 的共同作用实现肿瘤细胞靶向性,而不是单单依赖于GLUT1。又如,前文提及的前药29 ~32,结构相似但转运机制不尽相同[38]。前药29、31 和32 只作用于GLUT1,但葡萄糖修饰的前药30 同时作用于GLUT1 和OCT2。此外,来自若干不同课题组的研究结果也同样提示,有些糖基前药的作用机制并不依赖于GLUT。
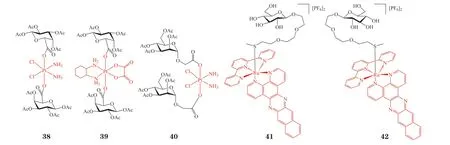
例如,Wang 等[43]合成了全乙酰化糖-Pt(Ⅳ)偶联物38~40,GLUT 抑制剂根皮苷对这些化合物的HepG2 细胞增殖抑制活性并无影响,说明这些化合物的细胞转运并不依赖于GLUT。Bonnet 课题组合成了葡萄糖修饰的多吡啶钌络合物41 和42,其结构中的葡萄糖分别为天然存在的D-葡萄糖和非天然的L-葡萄糖。由于D-葡萄糖可被GLUT 识别,而L-葡萄糖并非GLUT 的底物,通过比较化合物41 和42 的细胞毒性和细胞摄取可更科学地评价前药是否经GLUT 转运进入细胞。实验表明,化合物41 和42 在A549 细胞内均主要分布在线粒体,荧光强度并无差别,说明其细胞摄取不依赖于GLUT。细胞毒性实验显示,化合物41 和42经光照活化后对A549 和MCF-7 细胞的毒性相似。Bonnet 课题组推测化合物41 和42 的高脂溶性和所带的正电荷决定了其透膜行为和线粒体选择性分布,而与糖基无关[44]。
由此可见,准确地评价糖基对于前药靶向性的贡献至关重要。上述各实例中提到的方法,如考察GLUT 抑制剂对于前药细胞摄取和毒性的影响、GLUT 底物竞争实验、选择高表达和不表达GLUT的细胞进行对照、将D-和L-型糖作比较等方法应作为常规手段综合应用,并基于苷元即药物的特殊性质,科学系统地考察糖基对于前药靶向性的贡献,避免主观臆断的结论。
3.2 如何更精准地靶向糖转运蛋白和利用糖苷酶
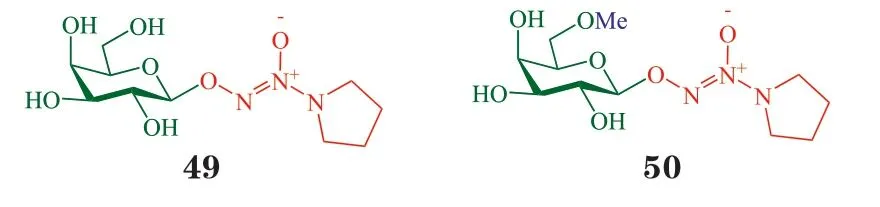
糖的结构多样性和糖基上的多羟基为前药设计提供了足够自由的空间,但同时也带来了复杂性和不确定性。从上文的实例可见,同样的药物,与不同的单糖偶联,抗肿瘤活性、靶向性等有时差异显著。而即便是同样的药物与同样的单糖偶联,连接在糖基的哪个位置也会显著影响前药的作用机制和抗肿瘤活性。这是一个相互影响、牵一发而动全身的复杂体系,因而需要更精准、更全面地设计药物。目前报道的糖基前药,绝大部分是将药物分子与糖基活性最高的端位羟基连接,与其他位置连接的相对较少,系统全面地比较与糖基C1~C6 位连接所得产物的活性的研究更为罕见。2016年,Lippard 课题组对此问题进行了非常出色的研究。课题组将同样结构的铂类通过同样长度的连接臂连接到葡萄糖的C1~C6 位,合成得到偶联物43~48,系统比较了上述偶联物的细胞摄取和细胞毒性。其中,将铂类药物与C1α位和C2 位连接所得的前药43 和45 更易在肿瘤细胞(DU145、A2780、SKOV3、A549)内富集。GLUT1 抑制剂cytochalasin B 对前药45 的细胞摄取影响最大,表明前药45 对GLUT1 的特异性依赖最高。相对应地,细胞实验证明,作用7.5 h后,前药45 对肿瘤细胞(DU145、A2780、A549)的毒性最大。用shRNA 部分敲除DU145 细胞的GLUT1后,前药45 对DU145 细胞的IC50升高近3 倍。上述研究揭示铂类与糖基C2 位连接所得的前药45 能更充分地利用GLUT 发挥靶向抗肿瘤的作用,也为今后糖基前药的设计提供了成功的示范[45]。当然,更精准、更全面的结构设计也对化学合成提出了更高的挑战。此外,结合计算机辅助药物设计方法预测不同位置修饰产物与GLUT 的相互作用,也为更精准的研究提供了有效的工具。释放出偶氮二醇盐阴离子,并进一步释放气体信使分子一氧化氮(NO)。然而,由于内源性β-半乳糖苷酶广泛分布于血液和多种组织,NO 释放的可控性不足,容易引起心跳加速、血压降低等毒副作用。课题组将半乳糖6 位的羟基替换为甲氧基得到化合物50,与原结构相比,该位置在空间上形成一个“凸起”。这一细微的结构改变使得化合物50无法被内源性的β-半乳糖苷酶识别。再通过酶定向改造,将β-半乳糖苷酶与底物结合口袋上的363 位组氨酸突变为丙氨酸后,空间上形成一个“洞”以容纳甲氧基。实验证实,改造后的半乳糖苷酶A4-β-GalH363A可特异性识别化合物50,水解底物后释放NO。动物实验中,将载有A4-β-GalH363A的凝胶注入裸鼠的后肢或肾脏,可实现NO 在该部位的特异性释放,用于后肢缺血的血管再生或治疗急性肾损伤。这种将基因工程改造的糖苷酶与前药相结合的设计,使得糖苷酶与糖基前药的相互作用可以得到更精准的调控,成功实现了药物在特定部位的选择性释放,大大拓展了今后糖基前药的发展范围[46]。
4 结语
糖苷酶是水解糖基前药并释放原药的关键,因此也决定着前药的肿瘤选择性,而当糖苷酶在肿瘤组织的分布特异性不足以实现靶向性时,就需要借助其他靶向配体(如叶酸等)实现靶向释药。2018年,Zhao 课题组另辟蹊径,通过对糖苷酶和糖基的改造实现了酶与底物的精准配对,提高了前药释放的选择性和可控性。化合物49 可经β-半乳糖苷酶水解
糖基用于靶向性抗肿瘤药物的前药修饰具有其独特的优势:首先,由于Warburg 效应,以GLUT 为代表的糖转运蛋白在多种肿瘤细胞表面过度表达,这就为利用各种糖基靶向肿瘤细胞提供了简单而有效的方法;其次,某些糖苷酶也在肿瘤组织中表达水平升高,利用这些糖苷酶水解糖基前药、释放原药,进一步提高了抗肿瘤药物靶向递送的可控性;此外,糖基修饰往往可以大幅提高药物的水溶性、改善其成药性。基于经典化疗药物如铂类、DOX、喜树碱、雷公藤甲素等的糖基前药设计方兴未艾。依托更为先进的前药设计理念,多靶点、多功能的糖基前药也正不断涌现。这些前药除了利用糖基外,还利用了多种肿瘤微环境的特征,如高表达的叶酸受体、过表达的OCT2 受体、高含量的还原性物质等,进一步提高了前药的肿瘤细胞靶向性。糖基前药也已被广泛应用于肿瘤之外的其他多种疾病,如威尔逊病和非酒精性脂肪肝的靶向药物递送,目前更多地集中在ASGP-R 介导的肝靶向递送。此外,目前基于糖基的纳米靶向给药系统正蓬勃发展。当然,随着该领域研究的不断深入,未来对科研工作者的要求也不断提高。如何准确地评价糖基对于前药靶向性的贡献?如何更全面地考察不同糖基、不同连接臂、不同连接方式对前药作用机制的影响?如何更精准地利用糖苷酶实现更特异性的前药活化?这些问题在今后的研究中值得思考,也对研究人员综合应用化学、生物学、药学等多学科技能开展研究的能力提出了更高的挑战。
