洋地黄毒苷的合成及基于抗肿瘤活性的糖基修饰研究进展
2020-08-10符登宪廖智文曾静万谦
符登宪,廖智文,曾静,万谦
(华中科技大学同济医学院药学院,湖北 武汉 430030)
1 洋地黄毒苷及其抗肿瘤活性
强心苷(cardiac glycoside)是一类具选择性强心作用的化合物,在动植物中均有分布。洋地黄毒苷(digitoxin,1)和地高辛(digoxin,2)均属于强心苷类。强心苷的结构中通常以甾体母核为苷元,其C17 位碳原子与不饱和内酯环相连,C3 位碳原子与糖基相连[1-2](见图1)。强心苷类药物发挥作用的机制通常被认为是通过抑制心肌细胞膜Na+/K+-ATPase 的活性,使细胞内的Na+增加、K+减少,最终使得心肌细胞内Ca2+水平增加,从而发挥正性肌力作用[3]。自1967年Shiratori 课题组[4]报道洋地黄毒苷等多种强心苷类化合物对肿瘤细胞的生长具有抑制作用以来,强心苷类药物的抗肿瘤活性也逐渐引起广泛重视,科学家们也对强心苷的抗肿瘤活性和作用机制进行了大量的探索[2,5-9]。
从洋地黄(Digitalis purpurea)中分离得到的洋地黄毒苷,是目前临床上治疗心力衰竭常用的强心苷类药物。洋地黄毒苷同样具有良好的抗肿瘤活性。已发现洋地黄毒苷对前列腺癌PC3、DU145和LNCaP 细胞株,乳腺癌MCF-7 细胞株,肾腺癌TK-10 细胞株,黑色素瘤UACC-62 细胞株,白血病K-562 细胞株,肺癌A549、NCI-H460 细胞株均具有良好的抑制活性[10-12];近年来,也发现洋地黄毒苷对胶质母细胞瘤[13]、肾细胞癌[14]、肝癌[15]具有一定治疗作用。另外,洋地黄毒苷的结构类似物地高辛对前列腺癌PC3、DU145 和LNCaP 细胞株,宫颈癌Hela 细胞株,肺癌A549、NCI-H460细胞株也显示出显著的抑制作用[10-12]。目前被广泛认可的洋地黄毒苷类化合物抗肿瘤作用机制可能是:其与肿瘤细胞Na+/K+-ATPase 亚基结合,进而影响后续的信号转导和细胞代谢,最终发挥诱导细胞凋亡的作用[16]。Lopez-Lazaro[17]、Newman等[10]、Prassas 等[8]、陈 果 等[18]、陈 大 朋 等[19]、Pongrakhananon[20]、Slingerland 等[21]与Schneider等[11]对洋地黄毒苷的抗肿瘤活性及其作用机制研究进展进行了详细综述。本文主要介绍近年来对洋地黄毒苷的合成以及针对其抗肿瘤活性进行的糖基结构修饰和构效关系研究的相关进展。
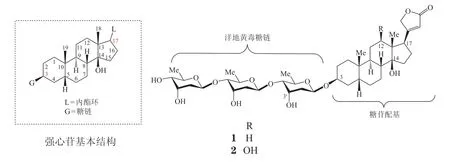
图1 强心苷的基本结构及洋地黄毒苷和地高辛结构Figure 1 Structure of cardiac glycoside, digitoxin and digoxin
2 洋地黄毒苷的全合成研究
强心苷类天然产物的重要生理活性引起了合成化学家广泛的兴趣。自Wiesner 等[22-23]于1985年首次报道洋地黄毒苷的全合成以来,McDonald 等[24-25]、Zhou 等[26-27]、Tanaka 等[28]相继开展了洋地黄毒苷的全合成研究。2012年,Heasley[29]对强心苷与相关天然产物的化学合成包括上述工作进行了总结和综述,罗列了历年科学工作者们在甾体母核和相关天然产物全合成中取得的进展。此后,Ma等[30]和Baryal等[31]又分别报道了2 种洋地黄毒苷的新的合成方案。
2011年,Ma 等[30]运用他们研发的金催化俞氏糖基化方法[32-36]顺利地在洋地黄毒苷元(3)上进行了糖基砌块的组装,通过九步反应以52%的总收率实现了洋地黄毒苷的构建(见图2)。在该合成工作中,以洋地黄毒苷元为原料,3 根糖苷键的构建都以端基离去基为邻炔基苯甲酸酯的洋地黄毒糖(4)为供体,以三苯基膦金(Ⅰ)三氟甲磺酸盐(Ph3PAuOTf)为催化剂。其中,供体4 以甲基吡喃葡萄糖苷为起始原料制备,其糖环C3、C4 位上具有较大立体位阻的叔丁基二苯基硅基(TBDPS)保护基,使得供体4 在进行糖基化时可以表现出良好的β-立体选择性。糖基的组装过程从洋地黄毒苷元的糖基化开始,通过从还原端向非还原端的连续糖基化实现:通过俞氏糖基化获得的化合物5 经由脱保护和C3 位羟基选择性乙酰化保护即得受体6,后续的二糖糖苷7 和三糖糖苷8 均可依此法进行制备。3 个糖的引入都利用相同的转化过程,很大程度上提高了合成的便利性和有效性。
2013年,Baryal 等[31]报 道 了 一 种Re(Ⅴ)催化的立体选择性合成β-洋地黄毒糖链(β-digitoxoside)的方法(见图3)。该方法以大位阻基团保护的6-脱氧-D-阿洛糖烯糖(6-deoxy-D-allal,9)为底物,在金属Re(Ⅴ)的催化下与受体10 直接发生烯糖糖基化,合成了二糖11。由于1,3-二直立键效应,该方法可以获得中等到优良的β-选择性。应用这一方法,Baryal 等首先合成了二糖(12)和三糖(13)。进一步应用还原锂化-消除反应顺利将三糖转化为烯糖(14)。但接下来烯糖14 的糖基化并没有再次应用Re(Ⅴ)催化的立体选择性糖基化方法引入洋地黄毒苷元,而是用了经典的三苯基膦氢溴酸盐(PPh3· HBr)催化的方法,以中等的产率获得了目标产物15,但遗憾的是,即使引入大位阻的叔丁基二甲基硅基(TBS)保护基,该步骤的糖基化选择性也不太理想。
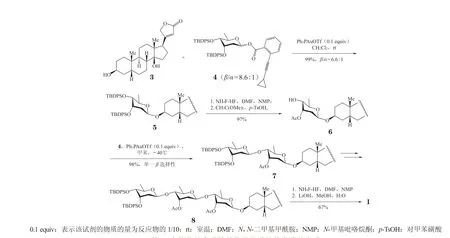
图2 金催化的俞氏糖基化用于洋地黄毒苷的合成Figure 2 Assembly of digitoxin by gold(I)-catalyzed Yu glycosylation
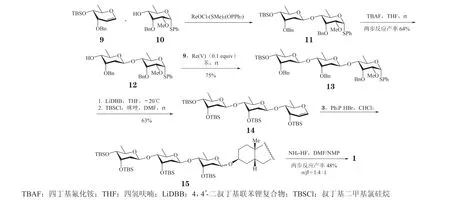
图3 利用Re(Ⅴ)催化的糖基化策略合成洋地黄毒苷Figure 3 Synthesis of digitoxin by Re(V)-catalyzed glycosylation
3 洋地黄毒苷的糖基修饰及改造
尽管洋地黄毒苷和地高辛具有广泛的抗肿瘤活性,但它们也有强心苷类药物常见的毒性大、治疗窗口窄等严重问题。为了寻找活性高、毒性低且能有效扩展治疗窗口的强心苷类抗肿瘤药物,近年来,药物化学家们对洋地黄毒苷类化合物进行了大量的结构修饰和改造研究,并对其构效关系进行了探讨。洋地黄毒苷的苷元部分被认为是药效基团,对药理活性起着关键的作用,因而针对苷元部分的结构改造对活性的变化起着直接的影响。但是苷元的改造通常都较为困难,因而报道不多,且大多以天然甾体的替换和细微的结构变化为主。Michalak 等[37]在2017年对强心苷类的苷元母核结构的合成和改造进行了总结和综述。另一方面,天然产物和药物的糖基化修饰往往能有效改变母体分子的理化活性,因而利用糖基随机化和糖基多样化的手段针对天然产物和药物进行糖基化修饰也成为了新药开发的有效手段。本文主要介绍利用化学方法对洋地黄毒苷的糖基部分进行的修饰和相应的构效关系研究。
3.1 钯催化糖基化和糖基化后修饰
O,Doherty 团队[38-42]在洋地黄毒苷及其衍生物的抗肿瘤活性的构效关系研究工作中取得了一系列研究进展。他们应用糖的从头合成法(de novocarbohydrate synthetic methodology)[43-45]和 糖 基 化后修饰(post-glycosylational modification)的策略,不仅完成了洋地黄毒苷全合成,也构建了一个包含不同糖结构单元的洋地黄毒苷类似物库(见图4)。该策略首先利用钯催化的糖基化策略,立体选择性地在α,β-不饱和吡喃酮(16)的C1 位引入洋地黄毒苷苷元得到化合物17,进一步对化合物17 的α,β-不饱和酮单元的羰基和碳碳双键进行选择性还原、环氧化或双羟基化等官能团化操作,多样性地引入各种糖基结构,得到化合物18a~18z。除了单糖外,重复应用该策略,也可以合成二糖和三糖的类似物。这些具备不同构型、不同取代形式和不同氧化程度的各种糖基取代基的洋地黄毒苷衍生物的合成,极大地推动了O,Doherty 等对洋地黄毒苷及其衍生物抗肿瘤活性构效关系的研究工作。运用该钯催化的糖基化和糖基化后修饰的合成策略,Elbaz 等[46]也于2012年合成了洋地黄毒苷的单糖衍生物18a,并对其抗肿瘤活性进行了较深入和细致的研究。
3.2 有机硼催化的区域选择性糖基化
2013年,Beale 等[47]利用他们开发的有机硼(20)催化的区域选择性糖基化方法,实现了洋地黄毒苷C4'''羟基的区域选择性糖基化(见图5)。该课题组以全乙酰化的C1 溴代糖19 为糖基供体,在有机硼催化剂20 和氧化银的体系中与洋地黄毒苷反应,以较好的产率和1,2-反式选择性得到各种四糖或五糖单元的洋地黄毒苷类似物(21a~21f)。其中,糖基化大多选择性地在位于平伏键的C4'''位发生,只有少数例外。但由于已经发现洋地黄毒苷的单糖类似物活性远强于多糖类似物,因而对这些化合物的抗肿瘤活性研究意义可能不大,目前也未见这些衍生物抗肿瘤活性研究的报道。
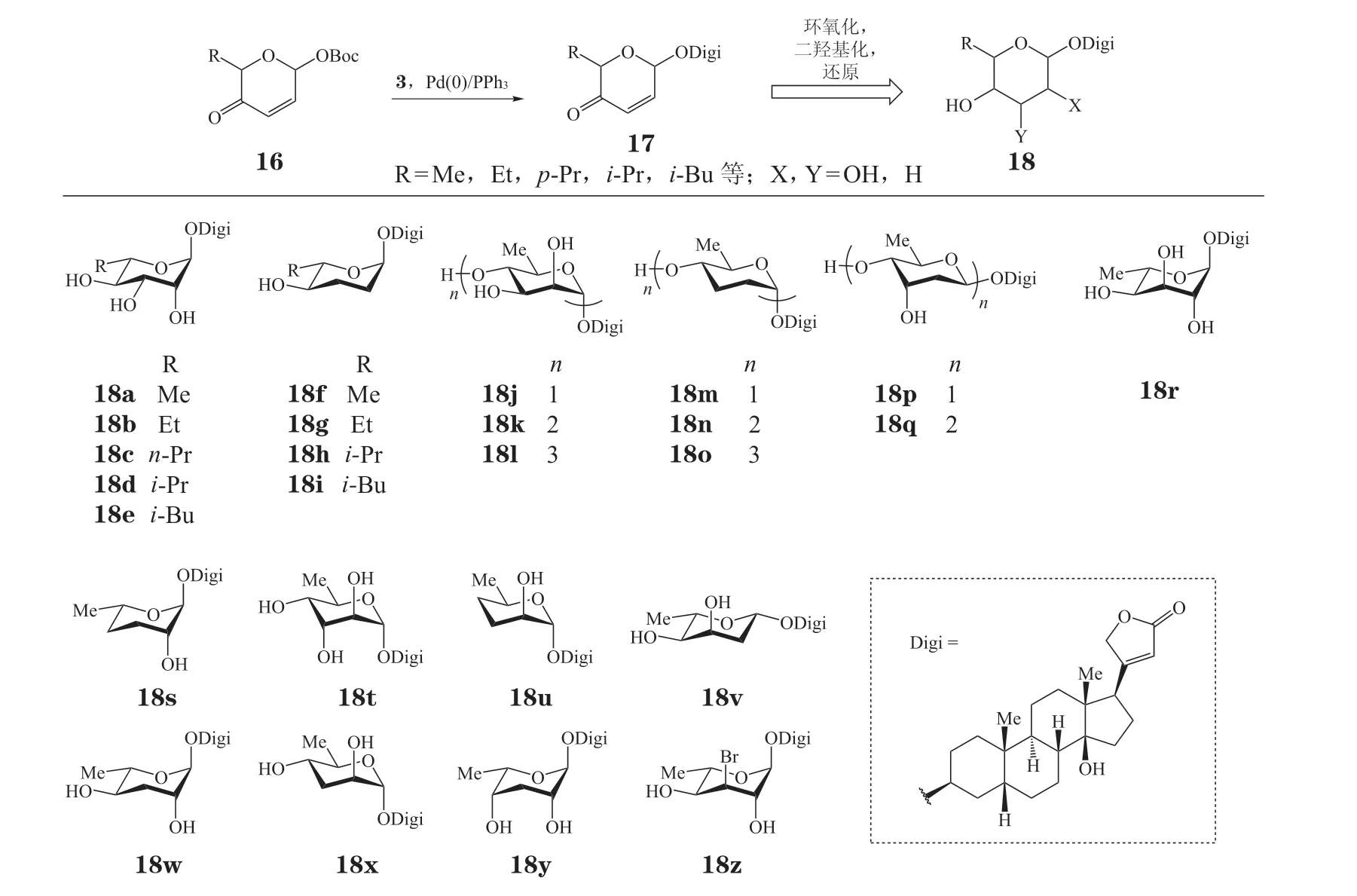
图4 利用钯催化糖基化及糖基化后修饰策略合成洋地黄毒苷类似物Figure 4 Synthesis of digitoxin analogues based on Pd-catalyzed glycosylation and post-glycosylational modification
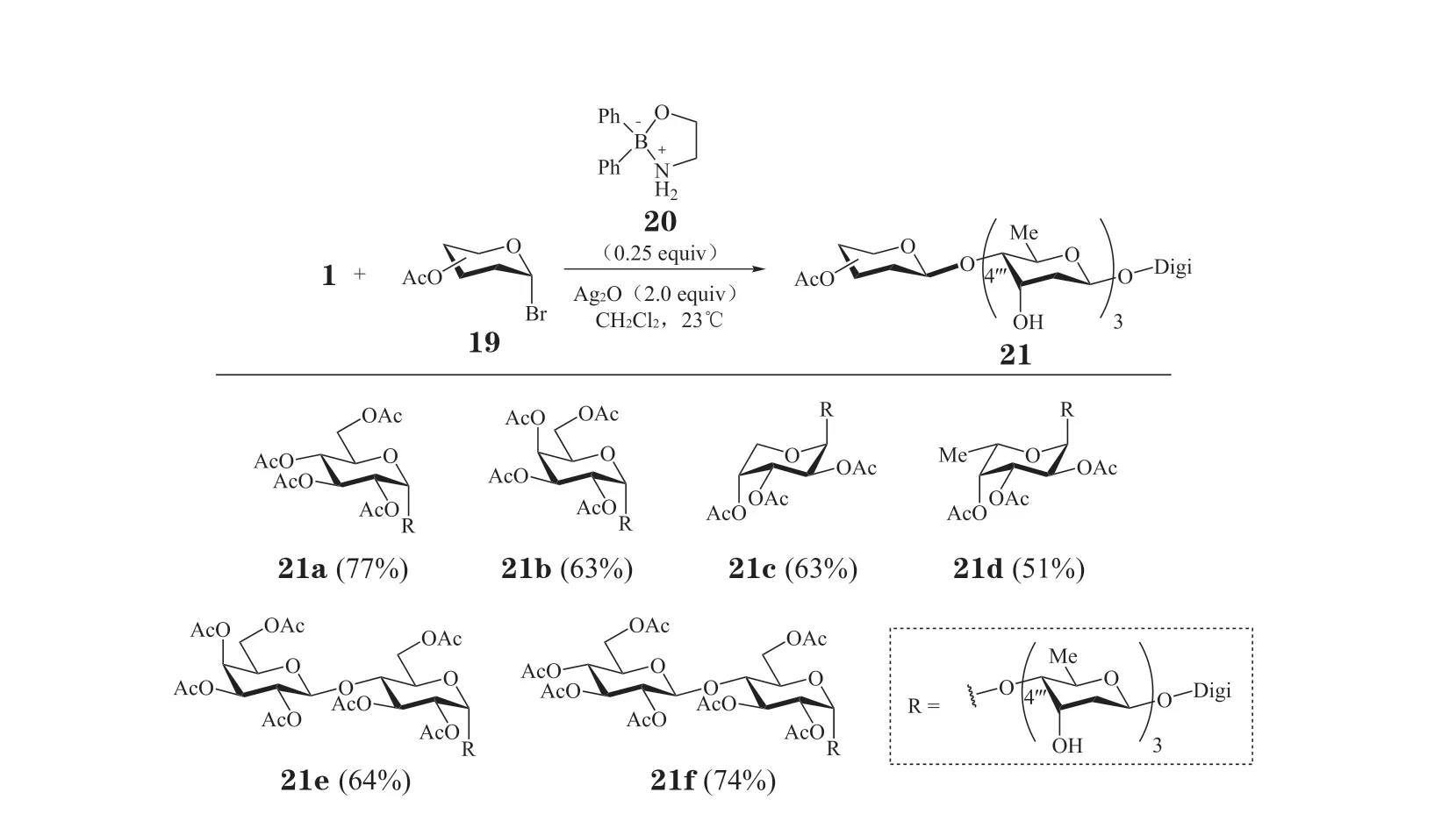
图5 应用有机硼催化剂对洋地黄毒苷进行区域选择性糖基化Figure 5 Regioselective glycosylation of digitoxin using organoboron-catalyst
3.3 金催化糖基化用于洋地黄毒苷的3-氨基糖修饰
3-氨基糖在抗癌药物和天然产物中广泛存在,其对此类化合物的抗肿瘤活性也具有重要的作用[48-51]。万谦及曾静等认为对洋地黄毒苷苷元进行3-氨基糖的修饰有可能提高化合物抗肿瘤活性并降低其毒性。该团队已利用3-氨基糖的多样化合成策略获得了多种天然的3-氨基糖及其衍生物[52]。在该工作基础上,他们进一步以端基离去基为邻炔基苯甲酸酯的3-氨基糖(22)为糖基供体,应用金催化的俞氏糖基化方法完成了洋地黄毒苷元的3-氨基糖修饰,获得了系列产物23a~23e[53](见图6)。在该方法中,尽管糖基化没有表现出明显的选择性,但是利用α-和β-异构体很容易分离的特点,同时获得了α-和β-构型的产物,有效地扩展了类似物库的多样性。应用该方法,万谦和曾静团队也对地高辛进行了3-氨基糖的修饰[53]。相关的抗肿瘤活性研究也在进行中。
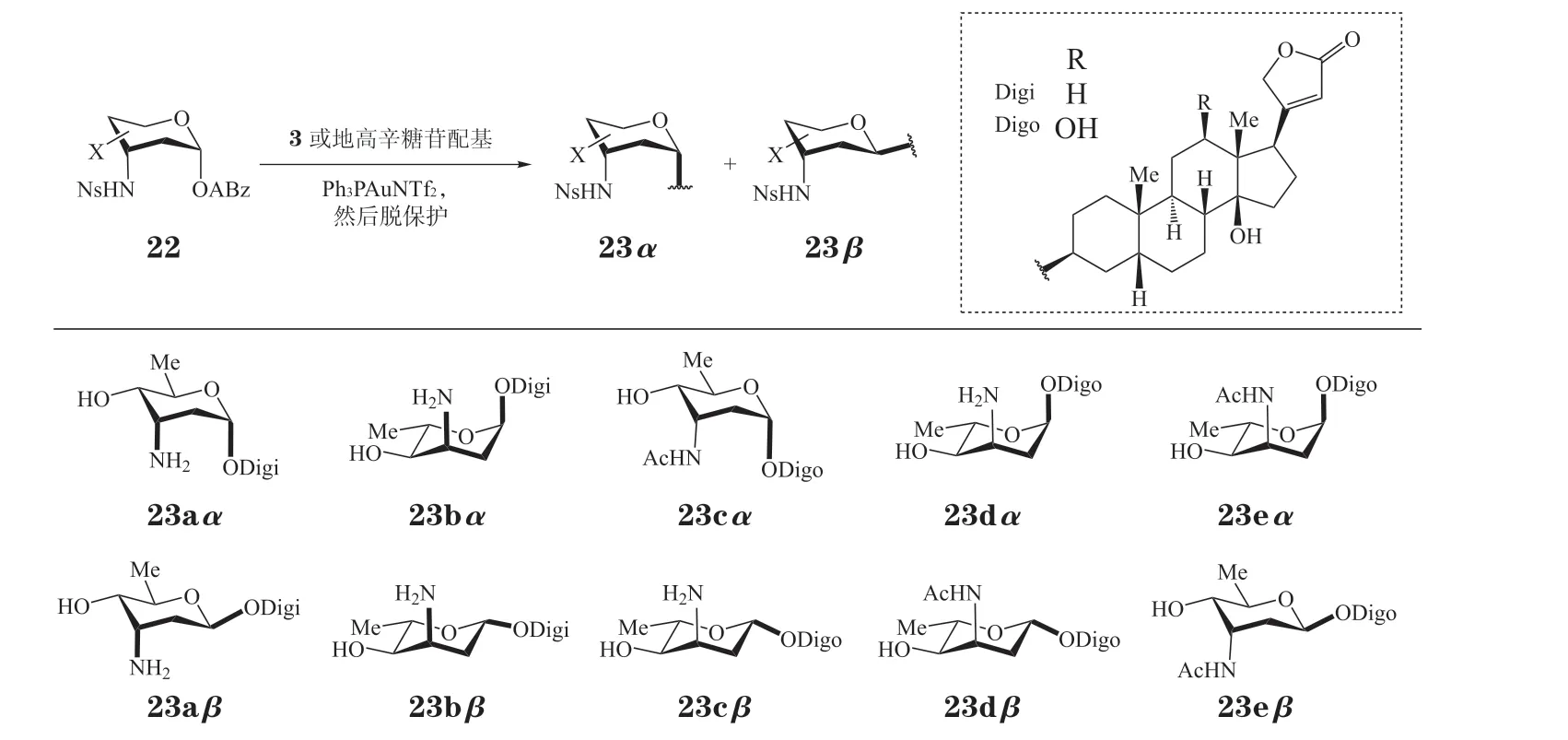
图6 洋地黄毒苷和地高辛的3-氨基糖修饰Figure 6 Structural modification of digitoxin and digoxin by 3-aminosugar
3.4 甲基羟胺-拟糖基化(MeON-neoglycosylation):洋地黄毒苷氮苷衍生物的合成
考虑到糖基化修饰的复杂性和挑战性,2005年,Langenhan 等[54]报 道 了 一 种 通 过 一 步反应在天然糖苷类药物苷元上引入糖基取代基的方法,他们将这种方法称为“拟糖基随机化(neoglycorandomization)”。该方法的核心在于利用还原糖24 和含O-甲基羟胺的苷元(25)缩合构建N-糖苷键,避免了糖基化反应的端基离去基的引入及活化,简化了糖基化反应的过程。基于拟糖基化的方法,O,Doherty 及其团队在2010年合成了洋地黄毒苷的单糖、二糖及三糖氮苷衍生物(26a ~26c),并对洋地黄毒苷氧苷和氮苷衍生物的抗肿瘤活性进行了对比[38]。
氨基糖的存在常常可以降低药物血脑屏障的透过性,因而有可能降低神经相关副作用。2015年,Zhang 等[55]利用拟糖基化方法合成了一系列洋地黄毒苷的氨基糖类氮苷衍生物,以对它们的抗肿瘤活性进行研究。如图7 所示,以叠氮取代的葡萄糖或木糖作为糖基化供体,通过拟糖基化和钯催化叠氮还原[56]两步反应得到系列糖环的C2'、C3'、C4'或C6'位被叠氮/氨基取代的衍生物(26d~26p)。运用拟糖基化合成新型氮苷衍生物的方法[54]和Koenigs-Knorr 糖基化方法,Wang 等[57]和Li 等[58]也对地高辛的氧苷和新型氮苷衍生物进行了合成,以此来研究它们的抗肿瘤活性和构效关系,并得到了一些抗肿瘤活性优于地高辛的衍生物。
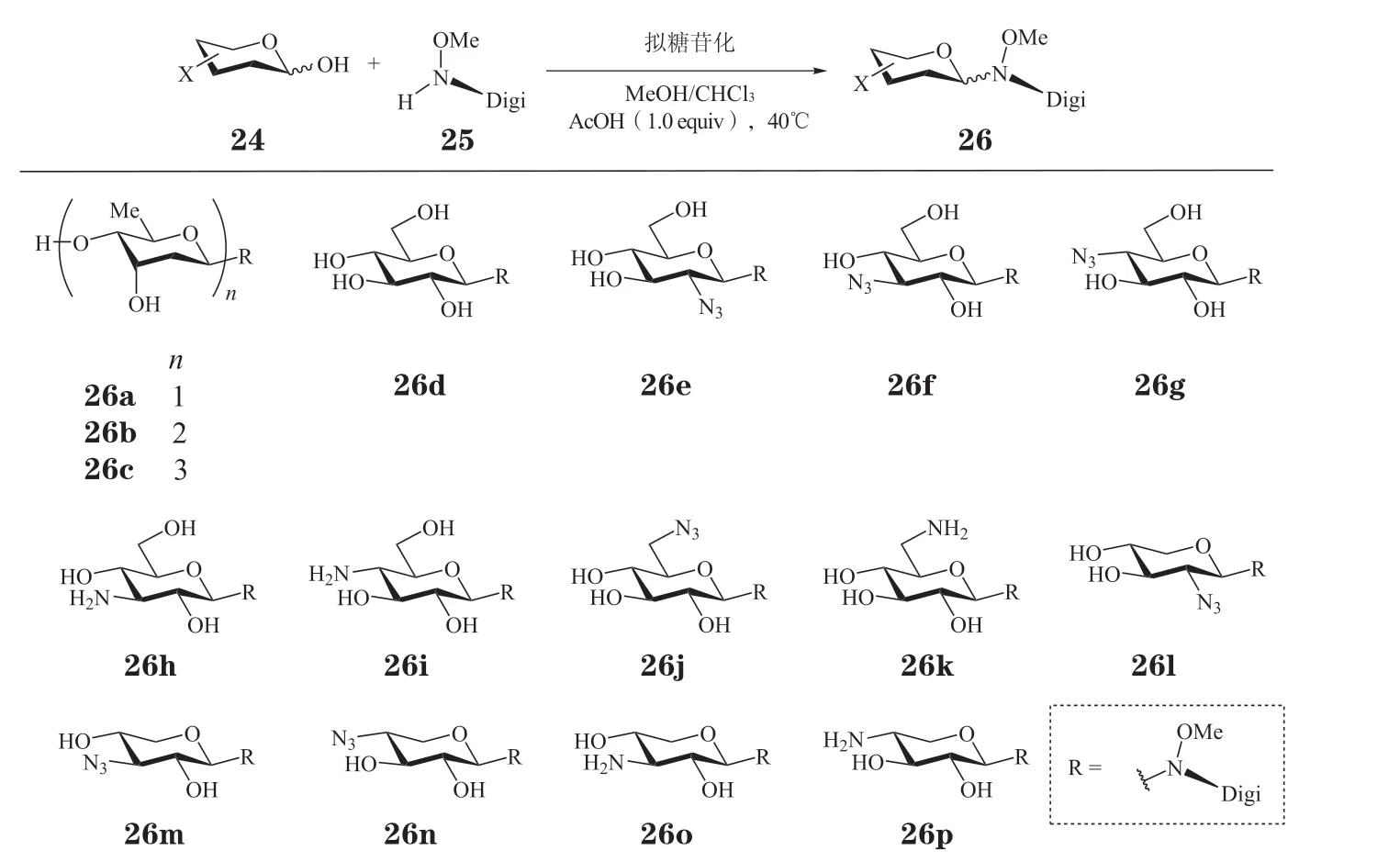
图7 拟糖苷化策略修饰洋地黄毒苷Figure 7 Modification of digitoxin by neoglycosylation strategy
3.5 含三氮唑和酰胺结构单元的洋地黄毒苷衍生物的合成
2019年,Boff 等[59]以洋地黄毒苷元为起始物,通过琼斯试剂氧化、硼氢化钠还原、对甲苯磺酰化、叠氮取代4 步反应得到了3-叠氮-3-脱氧洋地黄毒苷元(27),并以27 为起始物,合成了2 类洋地黄毒苷的衍生物。其中,他们运用点击化学(click chemistry)的方法,将炔丙醇苷糖基供体(28)在五水硫酸铜和抗坏血酸钠的作用下与27 进行糖基化偶联[60],继而将偶联产物29 进行脱乙酰化得到衍生物30(见图8A)。与此同时,Boff 等也尝试用非糖单元替换洋地黄毒糖单元:从27 出发,在三苯基膦作用下叠氮基团被还原成氨基[61-62],得到化合物31,再与氯乙酰氯反应生成关键中间体32[62-63];一方面,32 与乙醇胺反应生成衍生物33a,另一方面,中间体32 与碘化钾反应生成碘代物,然后在N,N-二异丙基乙胺(DIPEA)作为碱的体系中与相应化合物反应即可一锅法得到衍生物33b~33e(见图8B)[64-65]。
4洋地黄毒苷及其衍生物的抗肿瘤活性和构效关系
在上述洋地黄毒苷各类衍生物的合成工作的基础之上,研究人员对相应的衍生物的抗肿瘤活性进行了测试并对相应的构效关系进行了探究。通过他们的研究工作,洋地黄毒苷及其衍生物的抗肿瘤活性的构效关系逐步得到了进一步的阐明。1)甾体母核是洋地黄毒苷及其衍生物发挥药理活性必需的药效基团[66-67]。2)单糖衍生物表现出比二糖和三糖衍生物更高的活性[38-39],如单糖氧苷衍生物18p和单糖氮苷衍生物26a 的抗肿瘤活性分别高于相应的二糖和三糖氧苷(1,18q)、氮苷(26b ~26c)衍生物。3)氧苷衍生物的抗肿瘤活性高于前文所述异头位氮上有甲氧基取代的氮苷衍生物[38],如氮苷衍生物26a~26c 的抗肿瘤活性弱于相应的氧苷1、18q 和18p。4)糖基部分为L-构型的衍生物较D-构型的衍生物而言表现出更高的诱导细胞凋亡的活性,且糖基部分D-/L-立体构型的变化对诱导细胞凋亡活性的影响较C2、C3 位的取代造成的影响更为显著[39],如和相应的L-构型衍生物18a、18f 相比,D-构型衍生物18j、18m 诱导细胞凋亡的活性显著降低,同时18c 的活性显著高于18j,而18a 和18f 具有相当的生物活性。5)在洋地黄毒苷的单糖苷衍生物中,C2'直立羟基对衍生物抗肿瘤活性影响较小[42],如衍生物18w 和18f 的活性仅存在微小的差异,C3'羟基的存在对衍生物的抗肿瘤活性具有重要意义,但其立体化学(平伏羟基或直立羟基)对活性影响不大[42],如衍生物18a 和18r 具有几乎相同的抗肿瘤活性,而C3'脱氧的衍生物18w 和C3'羟基被更大的溴原子取代的衍生物18z 的抗肿瘤活性则更低。6)与C3'羟基相似,C4'羟基的存在对衍生物的抗肿瘤活性具有重要意义,但不同的是,C4'羟基的立体化学对衍生物的抗肿瘤活性的影响较之C3'羟基而言要大得多(平伏位羟基衍生物的活性比直立位羟基衍生物的活性高数倍)[42],如衍生物18w 的活性比18s 高约10 倍,比18y 高4~8 倍。7)C5'烷基取代的洋地黄毒苷单糖苷衍生物的抗肿瘤活性具有空间导向性,随着C5'取代基的空间位阻的增大,衍生物诱导NIC-H460 凋亡的活性也随之大幅地降低[41],如衍生物18a~18e、18f~18i,其抗肿瘤活性随C5'取代基位阻的增大而降低。8)在洋地黄毒苷氮苷衍生物中,3S-氮苷衍生物与3R-氮苷衍生物相比具有更显著的活性,同时拟糖苷-人类神经激肽A(neoglycoside-NKA)对接模型表明C3'-和C4'-氨基木糖取代的衍生物(26m~26p)具有更优良的抗肿瘤活性,其中,C3'氨基或叠氮取代的衍生物活性最佳(活性:26o ≈ 26m),C4'氨基取代亦可显著提高氮苷衍生物的抗肿瘤活性[55],如衍生物26j 的活性约为26d 的80 倍。9)Semose 及其团队[59]合成的两类洋地黄毒苷衍生物中,33a 和33b 具有最强的细胞毒性,它们的IC50值近似于作为阳性对照的抗癌药物紫杉醇,两者的结构中具有独特的羟基和乙醇胺基团,表明它们特殊的化学结构与其生物活性紧密相关;此外衍生物33a 和33b 的亲脂性较之其他衍生物更低,这种亲脂性特性在其与Na+,K+-ATP 酶的相互作用中发挥着重要的作用。在含三氮唑结构的衍生物中,三氮唑环上连接的甘露糖苷糖基单元上C1α-OH 和C2β-OH 的构型可能增强相应衍生物的活性,如衍生物30c 的活性高于30a、30b 和30d;同时相应糖苷衍生物糖基部分的羟基呈游离态时其抗肿瘤活性更高(相比乙酰基保护状态而言),如衍生物30a~30d 的活性高于29a~29d。就含酰胺结构的衍生物而言,缺电子的羰基碳原子上连接的基团的空间位阻增大时,化合物活性降低,且在酰胺羰基α位存在负电子的原子可降低衍生物的IC50值(与洋地黄毒苷元相比);如相比于33a 和33b,衍生物33c~33e 的活性显著更低。我们将部分构效关系总结如下(见图9)。
5 结语
洋地黄毒苷不仅具有优异的强心作用,同时也具有良好的抗肿瘤应用前景,进一步的研究还发现其类似物具有抗巨细胞病毒活性[68-69]、降低眼内压作用[70]、抗疱疹病毒活性[59]等生物学活性。由于洋地黄毒苷的毒性和狭窄的治疗窗口,近来人们研究的重点从天然洋地黄毒苷的合成转向具有更良好抗肿瘤活性的洋地黄毒苷衍生物的合成和相关的构效关系研究上,并取得了一系列具有指导意义的研究成果。洋地黄毒苷及其衍生物抗肿瘤活性的构效关系和作用机制仍未得到完全的阐明,有待更深入的探究。
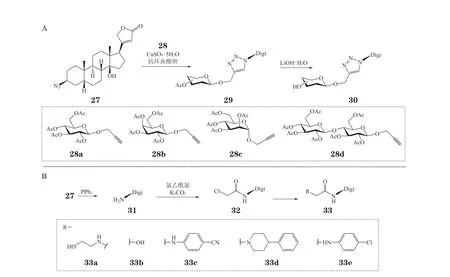
图8 含三氮唑和酰胺结构单元的洋地黄毒苷衍生物的合成Figure 8 Synthesis of digitoxin derivatives with special o-glycosyl trizol and amide fragments
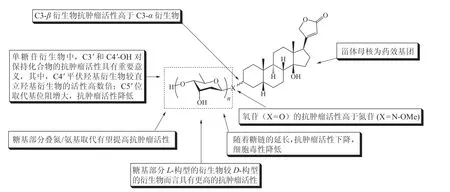
图9 洋地黄毒苷抗肿瘤活性的构效关系Figure 9 Structure-antitumor activity relationships of digitoxin
