理论预测Al3+与一种萘酚衍生物荧光探针分子形成配合物的结构
2020-07-28刘田田李文斌吴文鹏
刘田田,李文斌,潘 霄,吴文鹏*
(1. 河南大学 化学化工学院,河南 开封 475004;2. 宁波泰甬汽车零部件有限公司,浙江 宁波 315800)
铝元素在地壳中的含量排在第三位,仅次于氧和硅,广泛应用于人们的日常生活中.如果人体内积聚过量的铝,会引发多种神经退行性疾病[1-4].因此,多种分析方法被用来检测Al3+.其中,荧光探针分析法具有操作简便、灵敏度高、选择性好等特点,引起了人们的广泛关注[5-8].
最近,LIU等[9]用2-羟基-1-萘甲醛和苯甲酰肼合成了席夫碱型荧光探针分子L(结构式见图1),其最大吸收峰为359 nm;当加入Al3+后,最大吸收峰红移至402 nm. 由于激发态的C=N异构化(C=N

图1 L的结构式Fig.1 Chemical structure of L
isomerization)作用,探针分子L的荧光很弱,量子产率仅为0.002 8;当加入Al3+后,468 nm处的荧光增强了189倍.竞争性实验表明,L对Al3+有较好的选择性,L与Al3+的结合常数为4.8×105L/mol,检出限为8.87×10-7mol/L.通过工作曲线和质谱分析发现,L与Al3+的结合比为2∶2.
然而,文献[9]中未对L与Al3+形成的配合物的结构给出明确说明. 为了更深入地认识L与Al3+的相互作用,了解光谱变化的实质,根据之前的研究经验[10-11],我们将采用密度泛函理论详细研究L与Al3+的结合方式,推测L与Al3+形成的配合物的可能结构.
1 计算方法
在B3LYP[12-13]/6-31G**[14-15]水平下优化得到基态的几何结构,化合物的第一单重激发态的几何结构是在TD-B3LYP/6-31G**[16-17]水平上优化得到的,它们的垂直激发能和垂直发射能是在TD-M06[18-19]/6-31+G**[ 20-22]水平下计算得到.所有计算都是在Gaussian 09程序包[23]中完成的, 溶剂效应采用CPCM模型[24-25],溶剂为水.
2 结果与讨论
2.1 几何结构
对分子L我们设计出了2种可能存在的异构体(图2):L-N和L-T,前者羟基上质子未转移,后者质子转移到亚胺氮原子上.计算表明,后者能量比前者高0.28 eV.因此,前者更稳定.
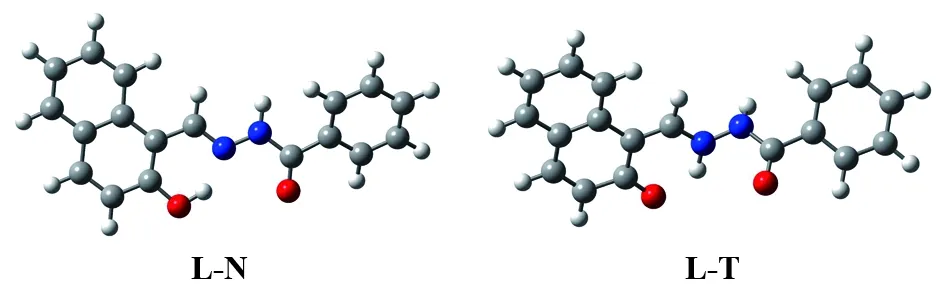
图2 化合物L的两种异构体的结构Fig.2 Two isomers of compound L
为了寻找L与Al3+相互作用时的可能作用位点,在L-N的几何结构的基础上,模拟得到了静电势(ESP),如图3所示.从图3中可以看出,L中两个氧原子附近的静电势较负,与文献[11]中1a类似.因此,两个氧原子是最可能的结合部位。

图3 L的静电势Fig.3 ESP of L
2.2 电子吸收光谱
2.2.1L的电子吸收光谱
模拟的L的电子吸收光谱 (考虑了20个激发态)如图4所示,相应的光谱数据列于表1中. 为方便比较,实验光谱[9]也一并画入图4中. 从图4中可以看出,尽管计算低估了高能激发态的吸收强度,但模拟得到的L的光谱与实验光谱峰的位置很接近.
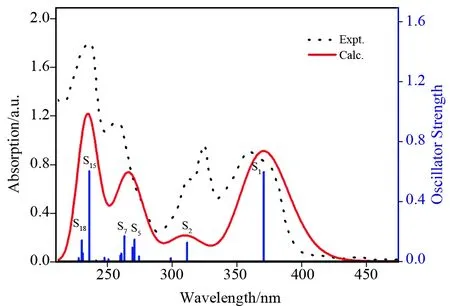
图4 模拟得到的L的电子吸收光谱Fig.4 Simulated electronic absorption spectrum of L

表1 化合物L的电子吸收光谱数据
2.2.2L-Al3+的电子吸收光谱
由实验文献[9]可知,探针分子L与Al3+形成2∶2型配合物. 根据图3中静电势的分布和实验上质谱峰的位置,我们设计并优化出了4种配位形式的化合物,它们都近似为蝴蝶形,如图5所示.其中,L-Aa、L-Ab为两个Al原子直接相连形成的化合物,L-Ac、L-Ad为两个铝原子通过两个氧桥键相连形成的化合物。它们均为同分异构体,可以通过计算它们的相对能量比较其相对稳定性.经计算,它们的相对能量分别为2.65、2.67、0和0.72 eV.所以,在这四种异构体中,L-Ac的能量最低,它是最有可能存在的配合物.计算发现,L-Ac的最大吸收波长在485 nm,振子强度为0.812 6.这与实验值402 nm相差较大,由此我们推断402 nm处的吸收不太可能是L-Ac产生的.
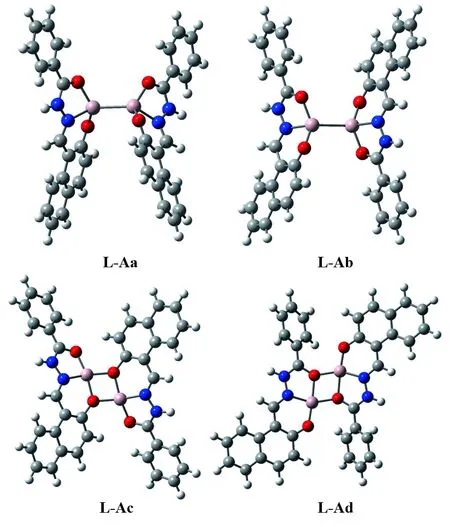
图5 L与Al3+可能形成的配合物的结构Fig.5 Possible structures of L-Al3+ complexes
考虑到阴离子可能会对L-Al3+的光谱产生影响,在L-Ac异构体的基础上引入Cl-,得到了结构L-Ae,如图6所示.

图6 L-Ae 的结构:(a) 俯视图 (b) 侧视图Fig.6 Structure of L-Ae (a) top view (b) side view
从图6可以看出,除了氯原子和两个苯环外,其余原子都在一个平面上,苯环稍微偏离了这个平面,与该平面的夹角大约为20°.该结构中,Al3+与三个氧原子、一个亚胺氮原子、两个氯离子形成六配位的结构;与萘环相连的氧原子形成氧桥,连接两个Al3+.
我们模拟得到了L-Ae的电子吸收光谱,如图7所示,相关数据列于表2中. 从图7和表2可以看出,尽管计算值低估了光谱的强度,但光谱峰的位置与实验[9]相符.因此,实验上的光谱有可能是L-Ae产生的.

图7 模拟得到的L-Ae的电子吸收光谱Fig.7 Simulated electronic absorption spectrum of L-Ae

表2 化合物L-Ae的电子吸收光谱数据
2.3 荧光光谱
为了确认形成的配合物的结构,我们优化得到了L-Ae的第一单重激发态(S1)的几何结构并模拟了L-Ae的荧光光谱,L-Ae的荧光主要来自于LUMO→HOMO的跃迁.最大发射波长在460 nm,振子强度为0.423 8,与实验光谱峰468 nm[9]一致.这进一步验证了L-Ae结构的正确性.
2.4 络合比的初步探究
为了解释Al3+与探针分子的络合比为2∶2,不是1∶1或者1∶2,我们优化得到了1∶1型(L-Af)和1∶2型(L-Ag)配合物的结构,如图8和图9所示. 其中,Al3+都采用六配位形式,L-Af中引入Cl-来满足六配位结构. 计算了Al3+与探针分子生成不同络合比的配合物的反应焓变(ΔH)和吉布斯自由能变(ΔG),结果列于表3中.从表3可以看出,L-Ae对应的ΔH和ΔG都是最负的,因此Al3+与探针分子L更倾向于生成2∶2型的配合物.
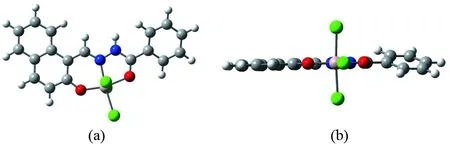
图8 L-Af的结构:(a) 俯视图 (b) 侧视图Fig.8 Structure of L-Af (a) top view (b) side view

图9 L-Ag的结构:(a) 俯视图 (b) 侧视图Fig.9 Structure of L-Ag (a) top view (b) side view

表3 不同络合比下计算的热力学数据 (单位: kcal/mol)
3 结论
用密度泛函理论优化得到了探针分子L的几何结构,用含时密度泛函理论模拟了其电子吸收光谱,理论光谱与实验光谱一致.进而筛选出了L与Al3+可能形成的配合物的几何结构,并通过电子吸收光谱和荧光光谱进行了验证.该结构中,每个Al3+分别与三个氧原子、一个亚胺氮原子、两个氯离子形成六配位的结构,与萘环相连的氧原子形成氧桥,连接两个Al3+.在此基础上,进一步研究了L与Al3+生成不同配比化合物的反应焓变和吉布斯自由能变.研究发现,L与Al3+的络合比为2∶2时反应焓变和吉布斯自由能变最负,最有可能形成.
