UPLC-PDA法测定甘草浸膏中甘草苷和甘草酸的含量
2020-07-16魏秀丽张传津冯言言李有志徐恩民
魏秀丽,张传津*,冯言言,崔 进,李有志,徐恩民
(1.山东省兽药质量检验所,山东省畜产品质量安全监测与风险评估重点实验室,动物源细菌耐药性监测与精准化用药山东省工程实验室,山东省动物用中药制剂工程实验室,济南 250022;2.齐鲁动物保健品有限公司,济南 250100)
甘草浸膏活性成分主要包括甘草苷、甘草酸,在人医临床用于治疗支气管哮喘、咽炎、支气管炎等;在兽医临床也用于缓解改善动物呼吸道疾病的症状,主要功效为祛痰止咳,也是制作甘草颗粒、甘草流浸膏等的原料。在中国药典和兽药典等文献中甘草浸膏或其他甘草制剂对甘草苷、甘草酸的含量测定均采用普通高效液相色谱方法[1-7],检测时间长。本实验研究了超高效液相色谱-二极管阵列检测法(UPLC-PDA)测定甘草浸膏中甘草苷和甘草酸的含量,简单快速,经济环保。
1 材料与方法
1.1 药品及试剂 甘草酸铵对照品,批号11073-2017020,含量97.7%,中国食品药品检定研究院。甘草苷对照品,批号111610-201005,含量94.9%,中国药品生物制品检定所。乙腈、冰醋酸、醋酸铵均为色谱纯;乙醇分析纯;超纯水。甘草浸膏,来自兽药生产企业报批产品。
1.2 仪器 Waters AcquityTMUltra performance LC超高效液相色谱仪,美国Waters 公司;Waters2695高效液相色谱仪,美国Waters 公司。
1.3 方法
1.3.1 供试品溶液制备 精密称取本品研细后的粉末,置具塞锥形瓶中,精密加入70%乙醇100 mL,密塞,称定重量,超声处理30 min,取出,放冷,再称定重量,用70%乙醇补足减失的重量,摇匀,滤过,取续滤液,即得。
1.3.2 标准储备液的制备 精密称取甘草苷对照品0.02012 g,置50 mL量瓶中,加70%乙醇使溶解并稀释至刻度,摇匀;精密称取甘草酸铵对照品(折合甘草酸为0.01959 g),置100 mL量瓶中,加70%乙醇使溶解并稀释至刻度,摇匀。
1.3.3 标准工作液的制备 取两种储备液适量,配置成混合对照品溶液:每1 mL含甘草苷200 μg、甘草酸20 μg的溶液。依次倍比稀释得一系列标准曲线工作液:10 μg/mL甘草苷100 μg/mL甘草酸、5 μg/mL甘草苷50 μg/mL甘草酸、2 μg/mL甘草苷20 μg/mL甘草酸、1 μg/mL甘草苷10 μg/mL甘草酸、0.5 μg/mL甘草苷5 μg/mL甘草酸、0.2 μg/mL甘草苷2 μg/mL甘草酸。
1.3.4 色谱操作条件及参数 色谱柱:T3色谱柱(2.1 mm×100 mm,1.8 μm);以乙腈为流动相A,0.1%冰乙酸+5 mmol/L醋酸铵为流动相B,按表1规定进行梯度洗脱;二极管阵列检测器,扫描范围190~400 nm,检测波长232 nm,柱温35 ℃,进样室温度10 ℃,流速为0.35 mL/min,进样量1 μL。
1.3.5 重复性试验和精密度试验 精密吸取甘草苷对照品溶液和甘草酸对照品溶液,制成每1 mL含甘草苷10 μg、甘草酸100 μg的混合对照品溶液,重复配置6份,进样,计算甘草苷、甘草酸峰面积RSD。
精密吸取甘草苷对照品溶液和甘草酸对照品溶液,制成每1 mL含甘草苷10 μg、甘草酸100 μg的混合对照品溶液,重复进样6次,计算甘草苷、甘草酸峰面积RSD。
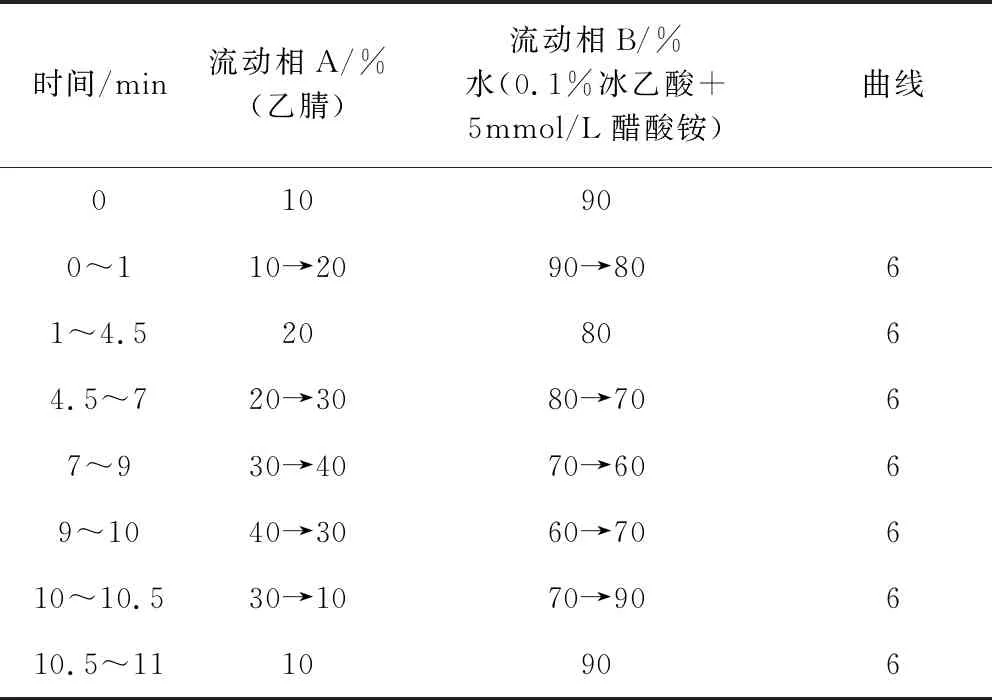
表1 梯度洗脱条件表Tab 1 Gradient elution condition
1.3.6 检测限和定量限的测定 对甘草苷、甘草酸的标准工作液进行UPLC分析,并逐级降低其浓度,以溶液检测时的信噪比(S/N)分别大于等于3和10 作为药物的检测限和定量限。
1.3.7 不同色谱条件样品检测比较 使用高效液相色谱仪(参照中华人民共和国兽药典2015年版甘草浸膏质量标准色谱条件[1], 流速1.0 mL/min;进样量10 μL)和超高效液相色谱仪在不同的色谱条件下运行,对同一样品中两主成分甘草苷、甘草酸的含量和出峰时间分别进行比较。
1.3.8 稳定性试验 进样室温度设置到10 ℃。取1.3.5项下10 μg/mL甘草苷100 μg/mL甘草酸溶液,在10 ℃放置3、6、12、24 h,各进样1 μL,计算甘草苷、甘草酸各自的峰面积RSD。
2 结果与分析
2.1 色谱分离 在优化的色谱条件下,测得甘草苷、甘草酸色谱峰峰形良好,且均达到基线分离,在1.3.4项条件下,甘草苷、甘草酸对照品和甘草浸膏样品待测液色谱保留时间为3.17、8.95 min左右,分离度均满足要求。
2.2 线性 在选定的色谱条件下,使用梯度洗脱的方法,可以有效地分离目的峰,甘草酸在10~200 μg/mL范围内的标准曲线:y=1167.9x+1986.4,R2>0.999;甘草苷在 1~20 μg/mL范围内的标准曲线:y=7348.3x+1104.8,R2=0.999。
理论塔板数:甘草酸、甘草苷均大于20000,分离度大于4.9,对称因子1.1。校正曲线见图1-图2。
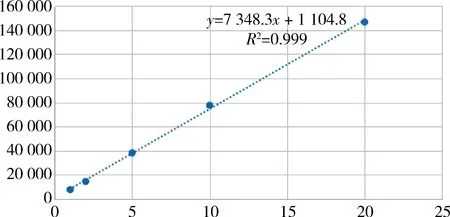
图1 甘草苷标准曲线Fig 1 Standard curve of liquiritin
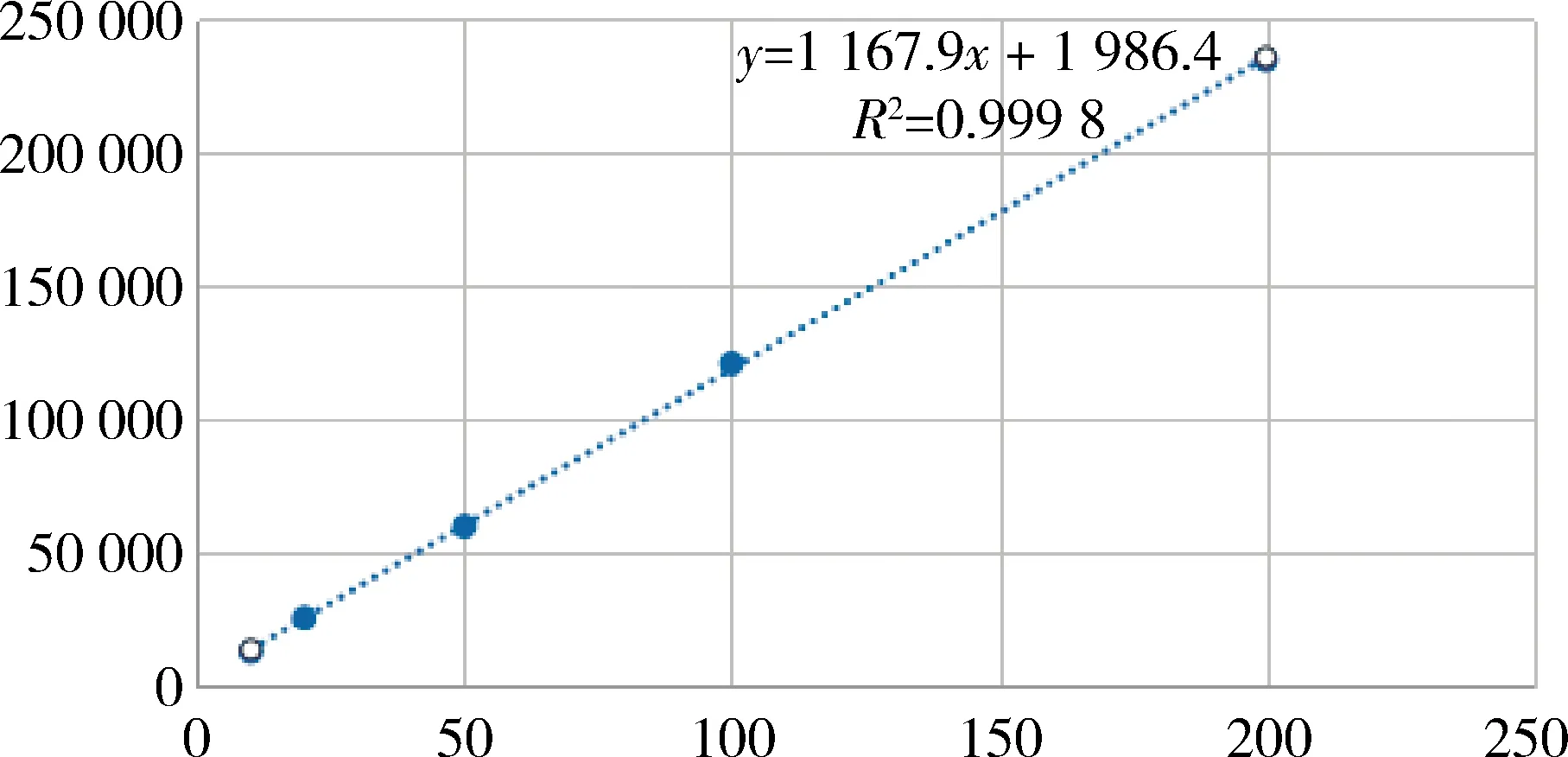
图2 甘草酸标准曲线Fig 2 Standard curve of glycyrrhizic acid
2.3 重复性试验和精密度试验 配制10 μg/mL甘草苷100 μg/mL甘草酸对照品溶液,重复配置6份,进样,计算甘草苷、甘草酸峰面积RSD分别为0.53%、0.48%,满足实验要求。
取10 μg/mL甘草苷100 μg/mL甘草酸对照品溶液,重复进样6次,计算甘草苷、甘草酸峰面积RSD分别为0.41%、0.39%,满足实验要求。
2.4 检测限和定量限 0.5 μg/mL甘草苷5 μg/mL甘草酸对照品溶液信噪比大于3为检出限,1 μg/mL甘草苷10 μg/mL甘草酸对照品溶液信噪比大于10为定量限。
2.5 不同色谱条件样品检测结果比较 不同波长下的主成分响应值的比较见表2。通过响应值比较及光谱图各成分的最大吸收波长,并顾及两种成分的峰面积平衡,选择232 nm。色谱图见图3-图13。
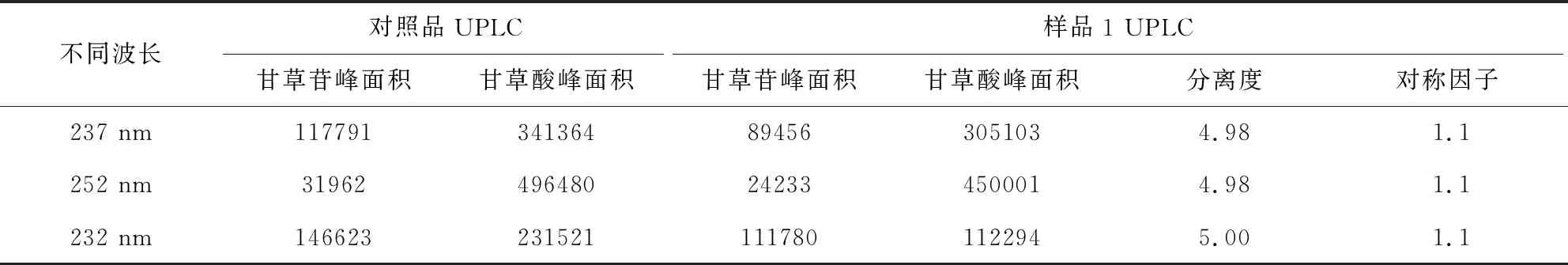
表2 不同波长下的主成分相应情况Tab 2 Principal component response at different wavelengths
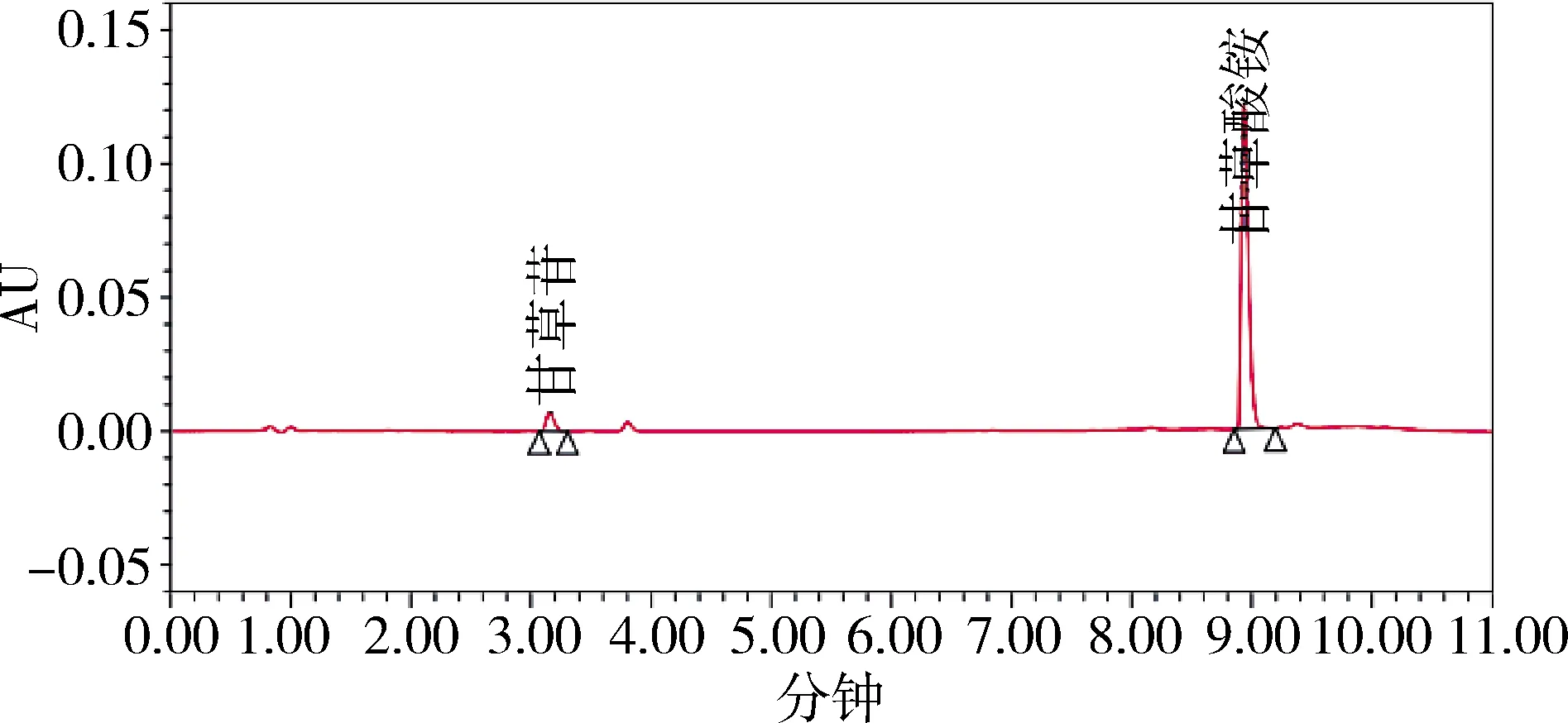
图3 对照品20μg/mL甘草苷200μg/mL甘草酸UPLC色谱图(252 nm)Fig 3 UPLC chromatogram of reference solution of 20μg/mL Liquiritin and 200μg/mL glycyrrhizic acid
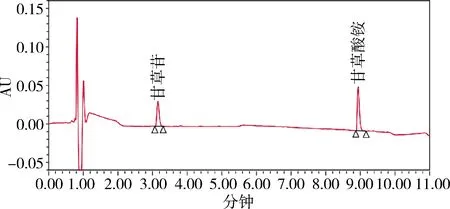
图4 对照品20μg/mL甘草苷200 μg/mL甘草酸UPLC色谱图(232 nm)Fig 4 UPLC chromatogram of reference solution of 20 μg/mL Liquiritin and 200 μg/mL glycyrrhizic acid
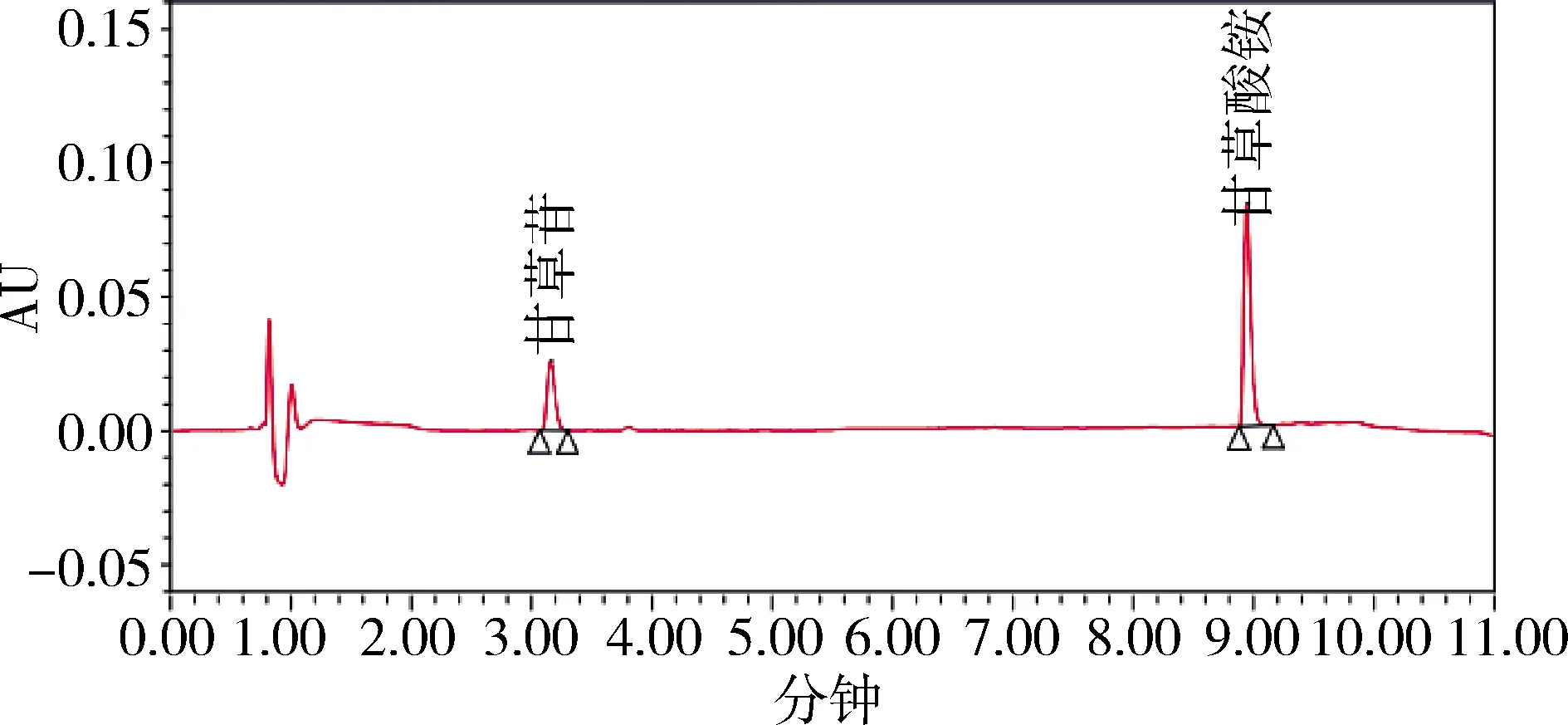
图5 对照品 20μg/mL甘草苷200μg/mL甘草酸UPLC色谱图(237 nm)Fig 5 UPLC chromatogram of reference solution of 20μg/mL Liquiritin and 200μg/mL glycyrrhizic acid
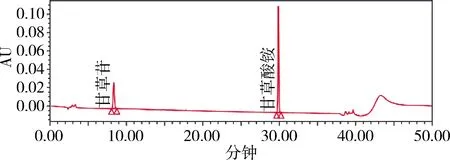
图6 对照品 20 μg/mL甘草苷200 μg/mL甘草酸HPLC色谱图(237 nm)Fig 6 UPLC chromatogram of reference solution of 20μg/mL Liquiritin and 200μg/mL glycyrrhizic acid
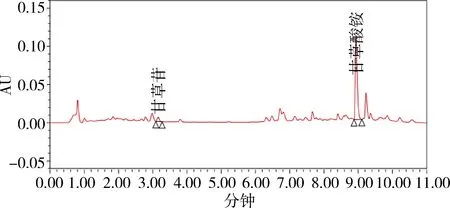
图7 样品1 UPLC色谱图(252 nm)Fig 7 UPLC chromatogram of sample 1
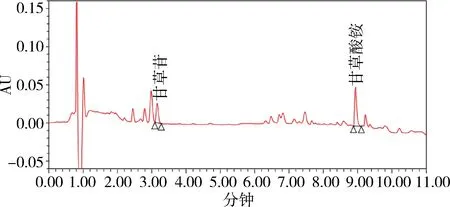
图8 样品1 UPLC色谱图(232 nm)Fig 8 UPLC chromatogram of sample 1
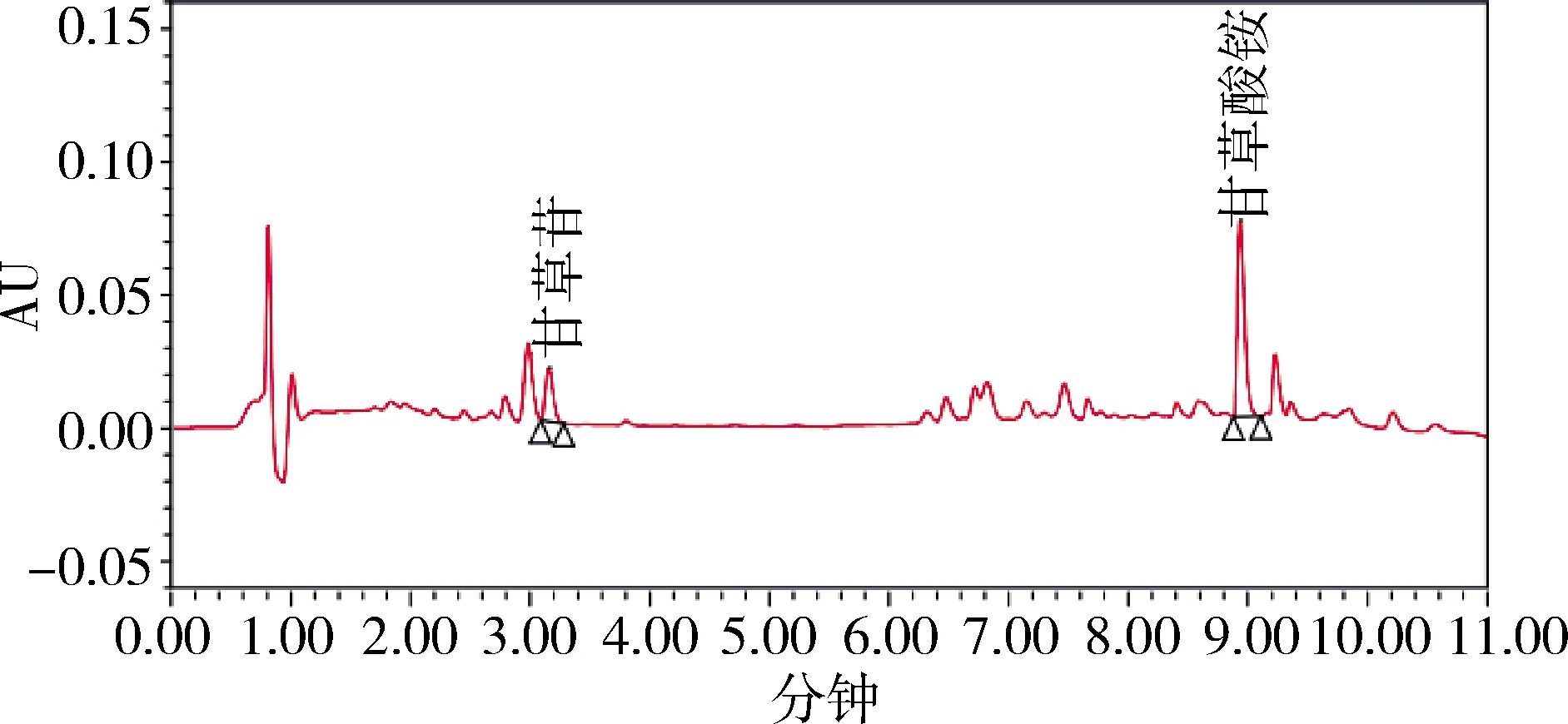
图9 样品1 UPLC色谱图(237 nm)Fig 9 UPLC chromatogram of sample 1
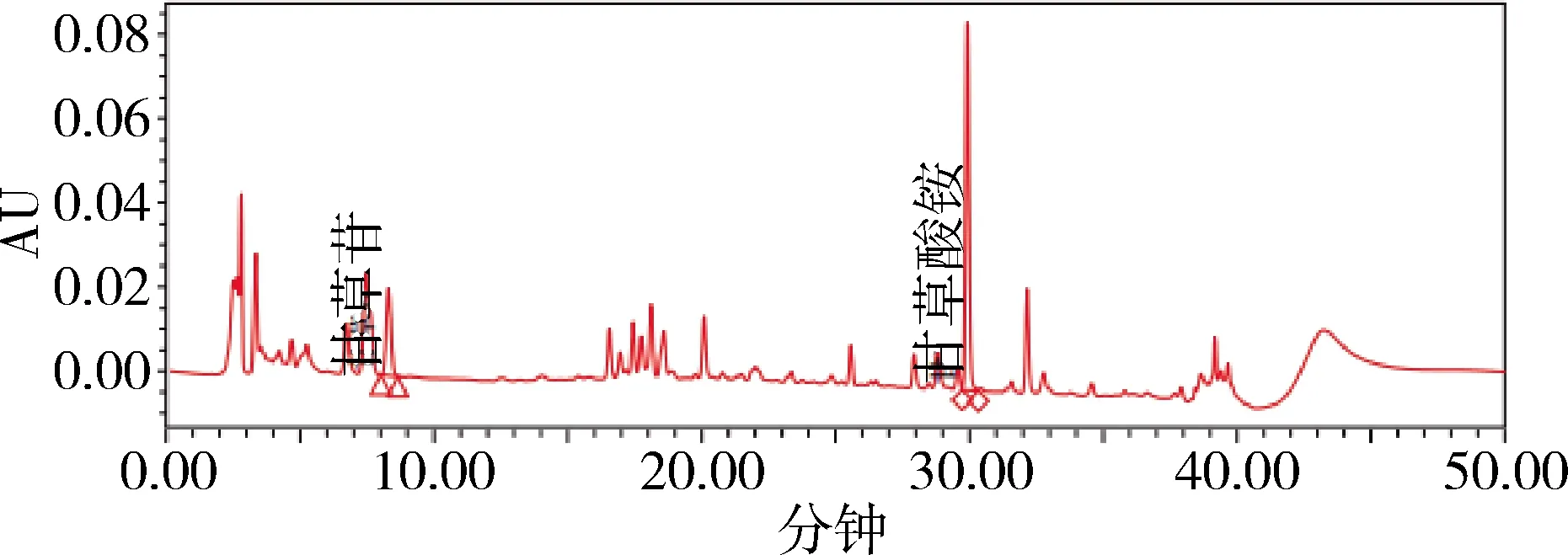
图10 样品1 HPLC色谱图(237 nm)Fig 10 HPLC chromatogram of sample 1
图11 样品2 UPLC色谱图(232 nm)Fig 11 UPLC chromatogram of sample 2
图12 样品2色谱图UPLC(237 nm)Fig 12 UPLC chromatogram of sample 2
图13 样品2色谱图UPLC(252 nm)Fig 13 UPLC chromatogram of sample 2
使用Waters超高效液相色谱仪(本实验色谱条件见1.3.4)和高效液相色谱仪(见1.3.7),对同一样品中按湿品计算两主成分甘草苷、甘草酸的含量分别进行了比较,无显著差异,相对偏差均小于1%,表明本实验方法简单可行,结果精确可靠。结果见表3。
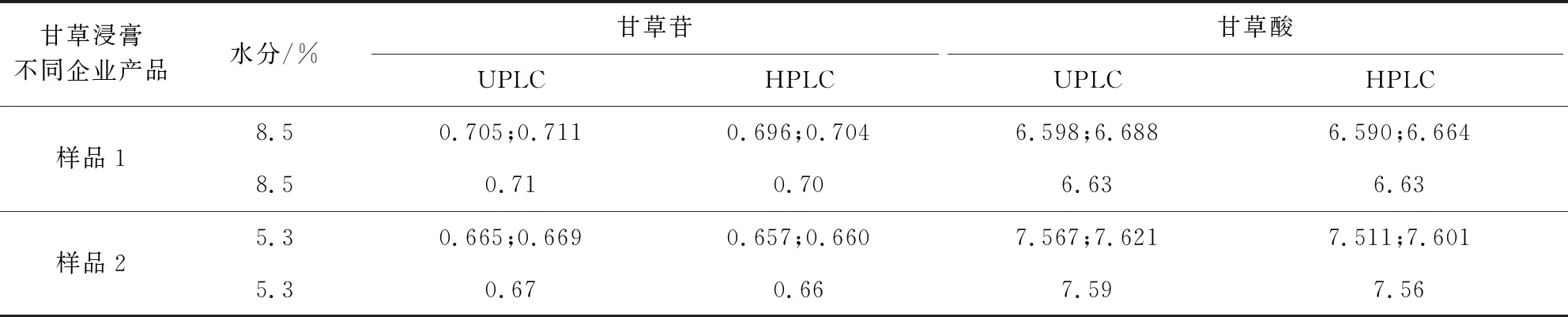
表3 不同色谱条件检测结果 (n=4)Tab 3 Test results using different chromatographic method (n=4)
由于两种色谱方法均已通过其方法学验证,均可以准确控制其指标成分。从检测效率上看,优选超高效法(UPLC),各色谱图见图3-图10。从样品噪音和峰形分离度等方面来看,优选超高效液相色谱仪条件,背景更干净,与杂质峰的分离度更好。
2.6 稳定性试验 按照实验方法,将对照品溶液贮存一段时间后进样,所得到的图谱中甘草苷、甘草酸峰面积RSD分别为0.38%、0.46%,说明样品稳定,满足实验要求。
3 讨论与结论
3.1 检测波长的选择 本研究将甘草苷、甘草酸的对照溶液利用PDA检测器采集其光谱图,在波长190~400 nm范围内进行扫描,发现甘草苷在232 nm处有最大吸收,甘草酸在252 nm处有最大吸收,根据光谱图可以判断含有主成分,能替代药典中2项鉴别:颜色反应和薄层鉴别,而且快速,不需要繁杂的处理过程,减少了硫酸、正丁醇、乙酸乙酯等多种提取溶剂和薄层展开溶剂,可以节约数小时提取、展开、喷雾等所需要的工作量。
参考兽药典含量测定项中选用的波长237 nm,在本实验中选择232、237、252 nm分别采集色谱图考察其峰面积响应、对称因子以及分离度等,由于甘草苷的响应值较低,最终选择232 nm作为检测波长。光谱图见图14-图15。两者保留时间分别为3.2、8.9 min。
3.2 流动相流速的比较 超高效液相流动相的流速为0.35 mL/min,需要运行11 min,每针运行需要3.85 mL流动相;高效液相色谱仪器流速为1.0 mL/min,运行时间50 min,每针运行需要50 mL流动相。超高效液相色谱更节约实验耗材溶剂等,每运行一针流动相节省46.15 mL。在废液处理高成本的今天,超高效液相色谱方法更有优势,更经济快捷。
3.3 本方法与药典方法的结果对比 本实验选择了两个企业的甘草浸膏进行含量测定,分别采用高效液相色谱方法和超高效液相色谱方法,甘草浸膏甘草苷和甘草酸含量相对偏差均小于1%,本方法运行一针需要11 min,比药典方法50 min节约了39 min的时间,大大提高检测效率。
本实验建立了超高效液相色谱法对甘草浸膏中甘草苷和甘草酸进行定性和定量测定,具备精准快捷、经济环保的特点,可以作为企业内控标准方法,有效控制甘草浸膏质量。
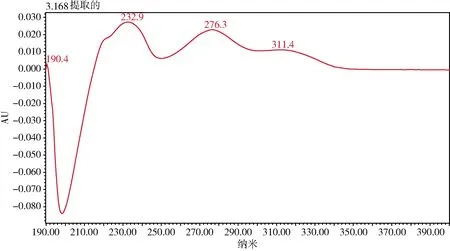
图14 甘草苷对照溶液光谱图Fig 14 Control solution spectrum of liquiritin
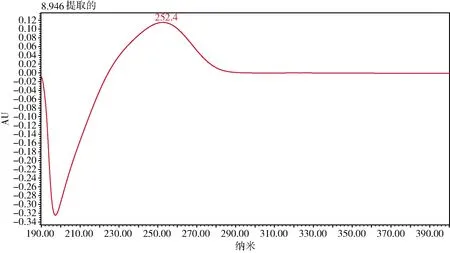
图15 甘草酸对照溶液光谱图Fig 15 Control solution spectrum of glycyrrhizic acid
