补肾解毒颗粒中4 种成分测定及HPLC 特征图谱建立
2020-07-08谭志滨徐俊鸿邓卓燕
谭志滨 徐俊鸿 邓卓燕 蓝 海
(广州中医药大学顺德医院, 广东 佛山528300)
补肾解毒颗粒是根据广东省名中医陈志雄教授验方补肾解毒方制成的制剂,包含熟地黄、山萸肉、山药、补骨脂、莪术、石上柏等药材,用于治疗血液系统恶性肿瘤,尤其对多发性骨髓瘤治疗有良效[1-6]。方中主药山萸肉所含以莫诺苷和马钱苷为主的环烯醚萜类成分具有抗炎止痛、免疫抑制等活性[7-8];补骨脂素、异补骨脂素为另一主药补骨脂的主要有效成分,对肉瘤和艾氏腹瘤等有着明显的抑制作用[9-11]。本实验通过HPLC 法测定补肾解毒颗粒中莫诺苷、马钱苷、补骨脂素、异补骨脂素含有量,并建立其特征图谱,以期为该制剂质量控制提供依据。
1 材料
熟地黄(批号181201,产地河南)、山药(批号190101,产地河南)、盐山茱萸(肉)(批号181201,产地浙江)、莪术(批号181201,产地广西)、石上柏(批号180901,产地广东)、补骨脂(批号181201,产地河南) 均购自广东天泰药业有限公司中药饮片厂,经广州中医药大学顺德医院徐俊鸿主任药师鉴定为正品,凭证标本保存于广州中医药大学顺德医院制剂中心。
莫诺苷(批号111998-201703,纯度97.4%)、马钱苷(批号111640-201808,纯度99.0%)、补骨脂素(批号110739-201617,纯度99.7%)、异补骨脂素(批号110738-201715,纯度99.5%) 对照品均购于中国食品药品检定研究院;甲醇、乙腈为色谱纯;其他试剂均为分析纯;水为超纯水。
Thermo U3000 高效液相色谱仪,配置TCC-3000SD柱温箱、LPG-3400SDN 泵、VWD-3100检测器、手动进样器(美国 Thermo 公司);ESJ200-4A 电子天平(十万分之一,沈阳神宇龙腾天平有限公司);FA2004 分析天平(常州市幸运电子设备有限公司)。
2 方法与结果
2.1 补肾解毒颗粒制备 称取熟地饮片500 g、山药饮片250 g、山萸肉饮片250 g、莪术饮片250 g、石上柏饮片250 g、补骨脂饮片250 g,置于提取容器中,加入12 倍量水煎煮2 h,滤过,收集滤液,滤渣加入10 倍量水提取1 h,收集合并滤液,滤液浓缩至相对密度1.25(55~60℃)。浓缩后,加入无水乙醇至含醇量50%~55%,静置12 h 以上,醇沉,滤过,收集滤液,浓缩至相对 密度1.10(55~60℃),浸膏、糊精、糖粉以1∶4∶1 比例添加糊精、糖粉等辅料,制 粒,干 燥,即 得(1 000 g)。
2.2 对照品溶液制备 精密称取各对照品适量,置于10 mL 量瓶中,50%甲醇定容至刻度,制成每1 mL 分别含1.0 mg 上述成分的贮备液,精密量取适量,置于同一25 mL 量瓶中,50%甲醇定容至刻度,制成每1 mL 含莫诺苷120 μg、马钱苷120 μg、补骨脂素50 μg、异补骨脂素80 μg 的溶液,即得。
2.3 供试品溶液制备 取颗粒适量研细,精密称取1 g,置于具塞锥形瓶中,精密加入50 mL 50%甲醇,超声(功率300 W、频率40 Hz) 处理30 min后取出,放冷至室温,50%甲醇补足减失的质量,摇匀,过0.45 μm 微孔滤膜,即得。
2.4 阴性样品溶液制备 按颗粒处方和工艺,制备缺山萸肉、缺补骨脂的阴性样品,按“2.3” 项下方法制备,即得。
2.5 色谱条件 Thermo Acclaim TM 120 C18色谱柱(4.6 mm×250 mm,5 μm);流动相乙腈-0.1% 磷酸,梯度洗脱(0~3 min,12% 乙腈;3~15 min,12%~18%乙腈;15~20 min,18%~25%乙腈;20~25 min,25%~38%乙腈;25~28 min,38%~40%乙腈;28~36 min,40% 乙 腈;36~40 min,40%~100%乙腈;40~50 min,100%~12%乙腈);体积流量1.0 mL/min;检测波长240 nm(0~25 min)、246 nm(25~50 min);柱温35℃;进样量20 μL。
2.6 方法学考察
2.6.1 线性关系考察 精密吸取对照品溶液0.10、0.20、0.50、1.00、2.00、5.00 mL 至5 mL量瓶中,50%甲醇稀释至刻度,在“2.5” 项色谱条件下进样测定。以溶液质量浓度为横坐标(X),峰面积为纵坐标(Y) 进行回归,结果见表1,可知各成分在各自范围内线性关系良好。
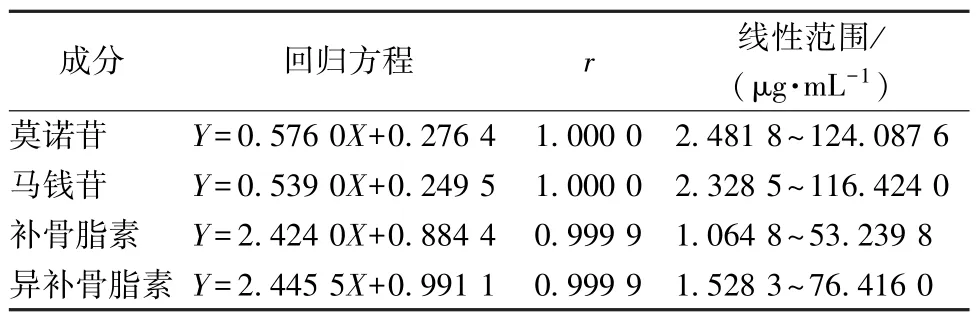
表1 各成分线性关系Tab.1 Linear relationships of various constituents
2.6.2 专属性考察 精密吸取对照品、供试品、阴性样品溶液各20 μL,在“2.5” 项色谱条件下进样测定,结果见图1。由此可知,供试品、对照品溶液均在相同保留时间出现吸收峰,阴性无干扰,表明该方法专属性良好。
2.6.3 精密度试验 精密吸取供试品溶液20 μL,在“2.5” 项色谱条件下进样测定6 次,测得莫诺苷、马钱苷、补骨脂素、异补骨脂素峰面积RSD分别为0.80%、0.42%、0.82%、0.75%,表明仪器精密度良好。
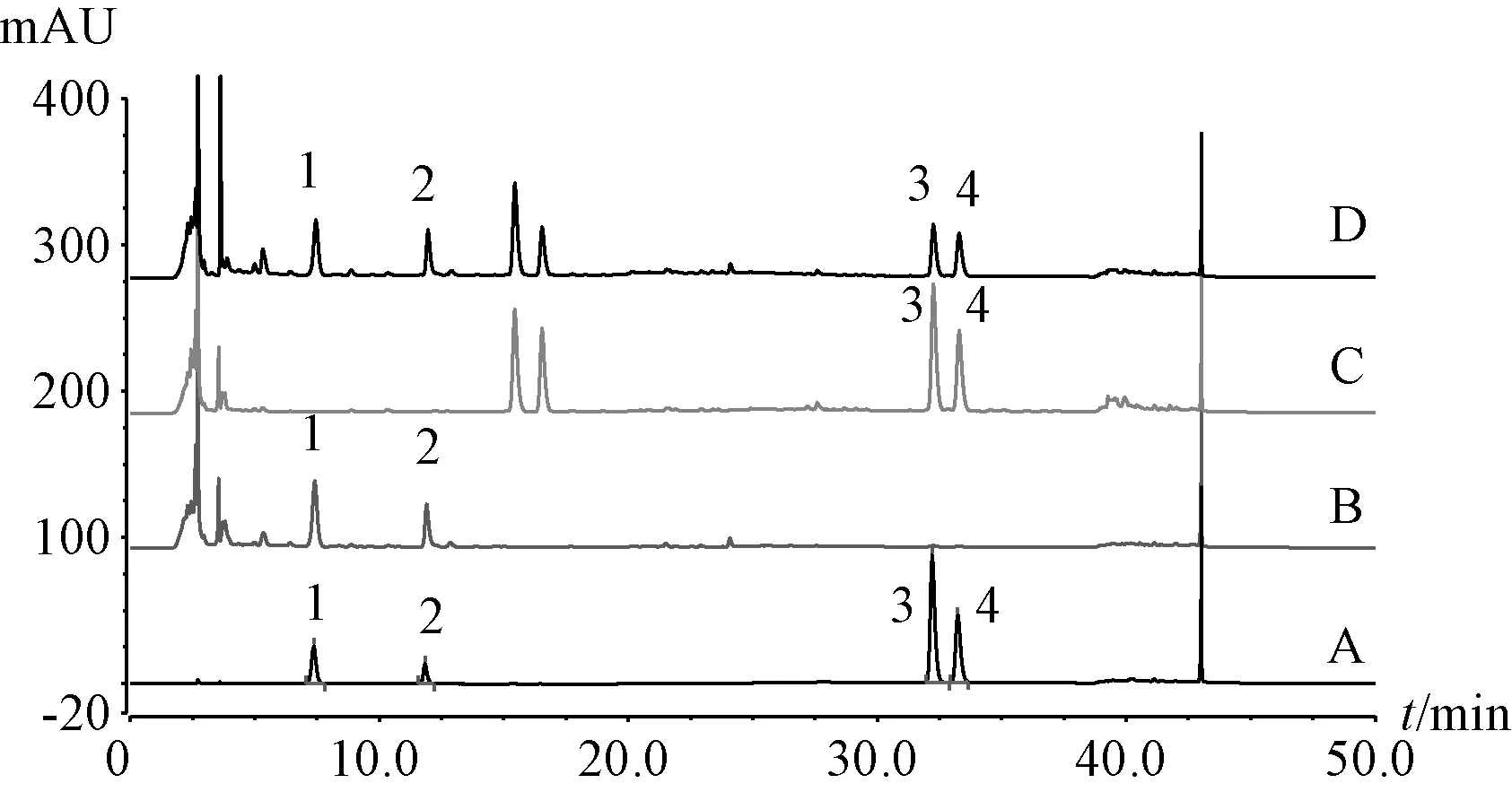
图1 各成分HPLC 色谱图Fig.1 HPLC chromatograms of various constituents
2.6.4 稳定性试验 精密吸取同一份供试品溶液,于0、1、2、4、6、8、24 h 在“2.5” 项色谱条件下进样测定,测得莫诺苷、马钱苷、补骨脂素、异补骨脂素峰面积RSD 分别为0.80%、0.61%、0.58%、0.59%,表明溶液在24 h 内稳定性良好。
2.6.5 重复性试验 精密称取同一批样品(批号190304) 6 份,按“2.3” 项下方法制备供试品溶液,在“2.5” 项色谱条件下进样测定,测得莫诺苷、马钱苷、补骨脂素、异补骨脂素含有量RSD分别为1.24%、1.00%、2.38%、1.58%,表明该方法重复性良好。
2.6.6 加样回收率试验 精密称取各成分含有量已知的颗粒6 份,每份0.5 g,按100%水平加入各对照品溶液,按“2.3” 项下方法制备供试品溶液,在“2.5” 项色谱条件下进样测定,计算回收率,结果见表2。
2.6.7 样品含有量测定 取15 批颗粒,每批平行2 份,按“2.3” 项下方法制备供试品溶液,在“2.5” 项色谱条件下进样测定,计算含有量,结果见表3。
2.7 HPLC 特征图谱建立
2.7.1 专属性考察 同“2.6.2” 项。
2.7.2 整体性考察 精密吸取供试品溶液20 μL,在“2.5” 项色谱条件下进样测定,并以最终梯度延长1 倍的时间进行洗脱,结果见图2。由此可知,在50 min 后无其他干扰峰,表明该方法整体性良好。
2.7.3 精密度试验 精密吸取供试品溶液20 μL,在“2.5” 项色谱条件下进样测定6 次,以补骨脂素为参照峰(S),测得各特征峰相对保留时间RSD 为0~0.23%,相对峰面积RSD 为0~1.42%,表明仪器精密度良好。

表2 各成分加样回收率试验结果(n=6)Tab.2 Results of recovery tests for various constituents(n=6)
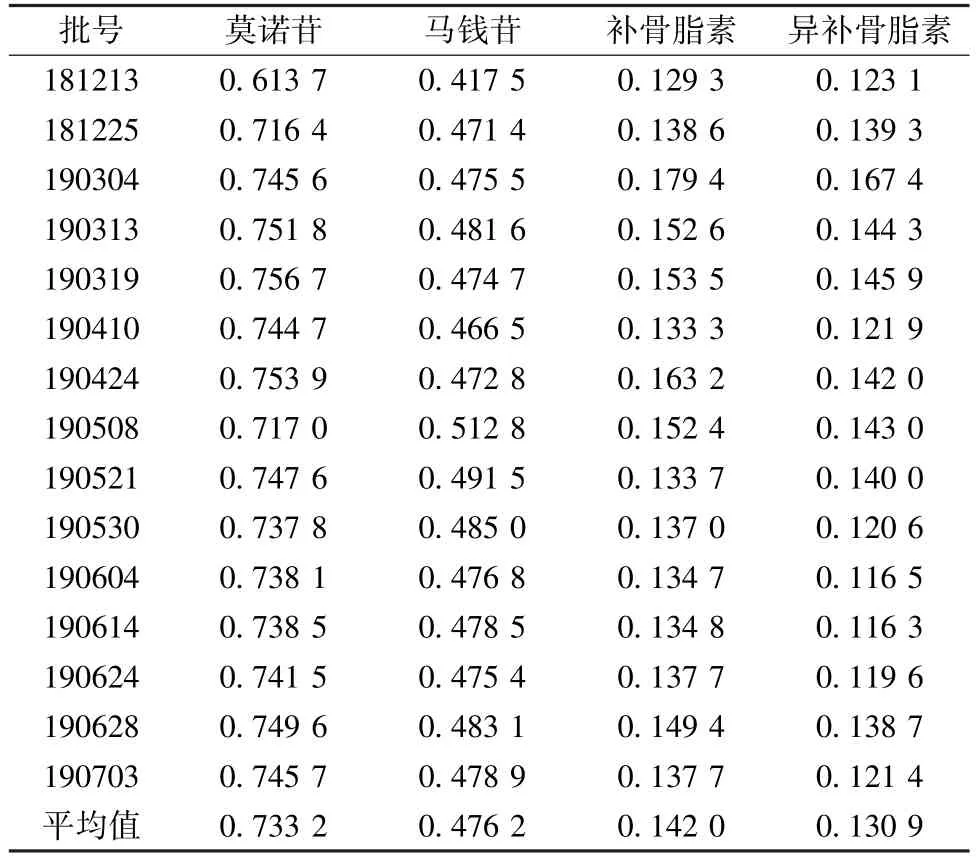
表3 各成分含有量测定结果(mg/g, n=2)Tab.3 Results of content determination of various constituents(mg/g, n=2)

图2 各成分整体性色谱图Fig.2 Integrality chromatogram of various constituents
2.7.4 稳定性试验 精密吸取同一份供试品溶液,于0、1、2、4、6、8、24 h 在“2.5” 项色谱条件下进样测定,以补骨脂素为参照峰(S),测得各特征峰相对保留时间RSD 为0~0.22%,相对峰面积RSD 为0~1.35%,表明溶液在24 h 内稳定性良好。
2.7.5 重复性试验取同一批颗粒(批号190304) 6 份,按“2.3” 项下方法制备供试品溶液,在“2.5” 项色谱条件下进样测定,以补骨脂素为参照峰(S),测得各特征峰相对保留时间RSD 为0~0.20%,相对峰面积RSD 为0~2.71%,表明该方法重复性良好。
2.7.6 图谱确定 取15 批颗粒,按“2.3” 项下方法制备供试品溶液,在“2.5” 项色谱条件下进样测定,以补骨脂素为参照峰(S),测定各特征峰相对保留时间、相对峰面积,建立特征图谱和对照图谱,结果见图3~4、表4。

图3 15 批样品HPLC 特征图谱Fig.3 Characteristic HPLC chromatograms of fifteen batches of samples

图4 各成分对照图谱Fig.4 Reference chromatogram of various constituents

表4 各特征峰相对保留时间、相对峰面积Tab.4 Relative retention time and relative peak areas of various characteristic peaks
2.7.7 计量学分析
2.7.7.1 相似度分析 利用中药色谱指纹图谱相似度评价软件(2012 年版),以液相图谱共有模式为参照图谱,将数据进行特征峰匹配,夹角余弦法计算相似度,结果见表5。由此可知,各批样品之间的差异较小,表明制备工艺稳定,重复性良好。

表5 15 批样品相似度Tab.5 Similarities of fifteen batches of samples
2.7.7.2 主成分分析 通过SPSS Statistics 19.0 软件进行主成分分析,结果见表6~7。由此可知,前2个主成分累积方差贡献率为87%,基本可以客观反映样品信息;补骨脂素、马钱苷对主成分1 的贡献度最大,而莫诺苷、马钱苷对主成分2 的最大。
2.7.7.3 聚类分析 通过SPSS Statistics 19.0 软件,以组间联接法、平方Euclidean 距离为测度,对15 批样品进行聚类分析,见图5。由此可知,当0<阈值<5 时,除批号181213 外其他样品聚为一类,批号181213 样品由于4 种成分含有量的差异而单独聚为另一类,结果与表5 一致,表明提取浓缩工艺稳定。

表6 主成分特征值和方差贡献率Tab.6 Principal component eigenvalues and variance contribution rates

表7 主成分分析结果Tab.7 Results of principal component analysis
3 讨论
3.1 色谱条件选择 本实验采用梯度洗脱,发现洗脱50 min 内时4 种成分分离效果良好,峰型对称,符合含有量测定要求。通过查阅相关文献结合VWD 紫外检测器参数考察所测成分紫外吸收,发现莫诺苷、马钱苷最大吸收波长均为240 nm[12-14],补骨脂素、异补骨脂素均为246 nm[15-16],为达到特征图谱信息量最大化和测定要求,检测波长选取240 nm(0~25 min)、246 nm(25~50 min)。流动相比较了乙腈-0.1%甲酸、乙腈-0.1%磷酸,发现以后者洗脱时各成分均实现基线分离,可达到定量分析要求。再进行体积流量、柱温等耐用性考察,发现分别在(1.0±0.1) mL/min、(35.0±1.0)℃范围均可重现,方法耐用性良好。

图5 15 批样品聚类分析Fig.5 Cluster analysis of fifteen batches of samples
3.2 提取条件优化 课题组前期研究发现,供试品中莫诺苷色谱峰峰型对溶剂要求较高,容易出现裂峰、峰型不对称,故选择2015 年版 《中国药典》 中的提取溶剂,即50%甲醇[17]。再对不同提取方式(超声、加热回流)、提取时间(20、30、40 min)、提取溶剂用量(50、25、10 mL) 进行考察,最终确定取样后精密加入50% 甲醇50 mL,超声(功率300 W、频率40 Hz) 处理30 min 作为最优提取条件。
3.3 HPLC 特征图谱分析 共指认了6个共有特征峰,其中峰1、2、S、6 分别为莫诺苷、马钱苷、补骨脂素、异补骨脂素,即为补肾解毒颗粒特征成分。再通过化学计量学评价各批样品相似度,发现不同批次之间差异较小,而且均采用同批药材,不存在原料差异,表明生产工艺稳定,各成分提取完全。
4 结论
本实验测定补肾解毒颗粒中莫诺苷、马钱苷、补骨脂素、异补骨脂素的含有量,并建立其HPLC特征图谱,再通过聚类分析和主成分分析对15 批样品进行化学计量学分析,发现该方法简便可靠,稳定性、重复性良好,可用于评价该制剂质量。
