非小细胞肺癌中MET抑制剂的耐药机制及应对策略研究进展*
2020-07-01
在非小细胞肺癌(non-small cell lung cancer,NSCLC)的治疗中,针对肿瘤驱动基因的靶向药物层出不穷。如口服小分子酪氨酸激酶抑制剂(tyrosine kinase inhibitors,TKIs)已被批准用于表皮生长因子受体(epidermal growth factor receptor,EGFR)突变、间变性淋巴瘤激酶(anaplastic lymphoma kinase,ALK)重排和ROS1重排的NSCLC[1-2]。间质-上皮细胞转化因子(mesenchymal-epithelial transition factor,MET)被认为是继EGFR、ALK 和ROS1 之后另一个重要的肿瘤驱动基因,针对MET 基因突变的靶向药物受到越来越多的关注。与MET基因相关的异常状态主要包括MET 14外显子跳跃突变、MET 基因扩增和蛋白过表达。其中,针对MET 14 外显子跳跃突变的靶向药物发展最快,已经上市和即将上市的药物包括克唑替尼、cabozantinib、沃利替尼、tepotinib、capmatinib等,另外还有许多药物正在进行临床研究[3-4]。但是,与其他靶向药物一样,MET抑制剂的耐药不可避免,给患者的治疗带来巨大的挑战。本文将结合MET基因异常的特点,重点对MET 抑制剂的耐药机制和应对策略进行综述,并提出未来MET 抑制剂的发展方向和面临的挑战。
1 MET基因突变
1.1 MET基因和HGF/MET信号通路
MET基因位于人类7号染色体(7q21-31),长度约125 kb,同时含有21个外显子[5]。由MET基因编码的蛋白为c-MET,也称为肝细胞生长因子受体(hepatocyte growth factor receptor,HGFR),是具有自主磷酸化活性的跨膜受体,属于酪氨酸激酶受体超家族,主要表达于上皮细胞。肝细胞生长因子(hepatocyte growth factor,HGF)是目前发现的唯一的c-MET配体,属于纤维蛋白溶酶原家族,主要表达于间质细胞。HGF能够与c-MET的细胞外结构域结合,促使c-MET发生二聚化、酪氨酸磷酸化,激活众多下游信号通路,如PI3K-Akt、Ras-MAPK、STAT和Wnt/β-catenin等,从而发挥促进细胞增殖、细胞生长、细胞迁移、侵袭血管及血管生成等效应。c-MET的结构和功能见图1。c-MET正常表达时促进组织的分化与修复,当存在异常时则可能促进肿瘤的增殖与转移[6]。HGF/MET信号通路异常激活主要包括MET 14外显子跳跃突变、MET基因扩增和c-MET蛋白过表达。

图1 HGF/MET信号通路及MET 14外显子跳跃突变
1.2 MET 14外显子跳跃突变
c-MET主要由E3泛素连接酶c-Cbl主导降解。MET 14外显子对应编码141个氨基酸,其所在的近膜结构域是c-MET的关键负性调控区,包含E3泛素连接酶c-Cbl酪氨酸结合位点(Y1003),参与c-MET蛋白的泛素化和降解。MET 14外显子的基因突变会引起14外显子跳读(exon skipping),使得含有E3泛素连接酶c-Cbl结合位点的近膜结构域缺失,进而导致c-MET蛋白泛素化障碍、c-MET稳定性增加和降解率降低,引起下游信号的持续激活,最终成为肿瘤的驱动基因。MET 14外显子跳跃突变的机制见图1。在NSCLC中,MET 14外显子跳跃突变的总体发生率为3%~6%[7-8],并且不与EGFR、ALK等NSCLC的其他驱动基因共存,提示其代表一种独立的肿瘤驱动基因[9-10]。但MET 14外显子跳跃突变可以与MET基因扩增和蛋白过表达并存[11]。
1.3 MET基因扩增
MET基因扩增即MET基因的拷贝数增加,包括整体染色体重复和局部区域基因重复[12],其中整体染色体重复是指肿瘤细胞中出现多条7号染色体。MET基因扩增通常伴有EGFR、KRAS等其他基因突变,有研究显示MET扩增可能并不是NSCLC的肿瘤驱动基因[13]。MET扩增与EGFR、KRAS等其他驱动基因的激活有明确的联系,可能是EGFR基因突变的NSCLC获得性耐药的机制之一。有研究显示,15%~20%的EGFR获得性耐药患者可检测到MET扩增[14-15]。另外,MET基因扩增往往提示NSCLC患者的预后较差[11]。
1.4 c-MET过表达或HGF过表达
HGF与c-MET结合可引起下游信号通路的激活,因此若存在c-MET或HGF过表达的异常情况,可能导致下游信号通路的持续激活,进而导致肿瘤的发生和发展。有研究表明,NSCLC中HGF/c-Met过表达与淋巴管生成密切相关[16]。c-MET过表达在肺腺癌中的发生率可高达65%,但其中仅10%的c-MET过表达伴有MET基因突变,因此c-MET过表达可能并不是原发致癌驱动因素,更可能是作为其他驱动基因激活后产生的二次事件,从而促进肿瘤的生长[17]。
2 MET抑制剂的作用机制
基于HGF/c-MET 信号通路异常激活,MET 抑制剂成为NSCLC 的重要治疗手段。跟据HGF/c-MET信号通路中作用位点的不同,可将MET抑制剂分为3大类:抗HGF单克隆抗体、抗c-MET单克隆抗体和小分子TKI。前两者分别在细胞外与HGF 和c-MET 结合,从而阻止HGF与c-MET的结合及受体磷酸化,阻止信号传导;小分子MET-TKI 作用于膜内催化域从而阻止蛋白磷酸化,阻断信号传导(图1)。目前研究最多且最具有治疗潜力的是小分子MET-TKI。
2.1 MET-TKI
MET-TKI可分为3种类型(Ⅰ型、Ⅱ型和Ⅲ型)[18]。Ⅰ型TKI是ATP竞争性抑制剂,与MET主链中的氨基酸残基形成氢键,其中又分为Ⅰa型和Ⅰb型,Ⅰb型TKI可以结合的位点较少,不包括甘氨酸残基的G1163位点(类似于ALK的G1202和ROS1的G2032位点),因此特异性较高。临床常用的药物克唑替尼属于Ⅰa型METTKI,tepotinib、沃利替尼和AMG337等均属于Ⅰb型METTKI。Ⅱ型MET-TKI一般为多靶点TKI,不仅作用于ATP结合位点,还能通过管家基因突变进入非活性DFG-out构象形成的疏水口袋,对产生二次突变的MET仍具有抑制作用,或许可以逆转由Y1230等突变引起的Ⅰ型MET-TKI耐药[3]。cabozantinib属于Ⅱ型MET-TKI。Ⅲ型MET-TKI作用于与ATP结合位点完全不同的变构位点,目前尚无药物进入临床研究阶段。
2.2 抗HGF/c-MET单克隆抗体
该类药物的原理是通过单克隆抗体竞争性结合HGF 或c-MET,阻断HGF 和c-MET 之间的相互结合。此类药物包括rilotumumab(AMG-102)、ficlatuzumab(AV-299)和TAK-701 等[19-21]。抗c-MET 单克隆抗体与c-MET 结合,一方面阻断了HGF 与c-MET的结合,另一方面此类受体与配体在细胞表面的结合不引发c-MET 信号传导,无法诱导c-MET 二聚化。遗憾的是,部分抗HGF/c-MET 单克隆抗体的临床试验失败,部分仍在进行中,目前尚无成功的抗HGF/c-MET单克隆抗体药物上市。
3 MET抑制剂的耐药机制
MET 抑制剂的耐药机制研究主要集中在METTKI药物,可分为原发性耐药和继发性耐药。
3.1 原发性耐药
MET-TKI通过特定氨基酸上的疏水相互作用而与c-MET的ATP口袋紧密结合。在MET作为致癌驱动因素的肿瘤中,用MET-TKI进行的体外诱变分析已鉴定出针对Ⅰ型MET抑制剂的几种优势耐药突变(Y1230,D1228)和部分轻微耐药突变(F1200,V1155)[22-23]。
Fujino 等[24]对8 种MET 抑制剂进行的体外试验研究显示,MET基因中D1288和Y1230突变可能导致Ⅰ型MET-TKI耐药,L1195和F1200突变可能导致Ⅱ型MET-TKI 耐药,但Ⅰ型和Ⅱ型MET-TKI 之间无交叉耐药现象。另外,有研究显示HGF/MET 的下游信号通路改变,如PI3K 信号通路的改变,是MET 抑制剂原发耐药的机制之一[25]。
3.2 继发性耐药机制
在应用MET抑制剂初始治疗有效后,可能出现新的MET结构域改变,从而导致继发性耐药的发生。Dong等[26]报道1例MET 14外显子突变的NSCLC患者在应用克唑替尼治疗后耐药,二代测序显示外周血循环肿瘤细胞同时出现MET基因D1228N/H和Y1230H突变。另有研究报道MET 14外显子突变的NSCLC患者接受克唑替尼治疗后,产生获得性耐药突变MET D1228N和MET Y1230C[27-28]。Bahcall等[29]研究显示MET基因中D1228V点突变可能是沃利替尼继发性耐药的原因之一。韩森等[30]报道了1例MET 14外显子突变的晚期肺肉瘤样癌患者应用沃利替尼治疗后出现耐药,二次活检提示新出现了成纤维细胞生长因子受体(fibroblast growth factor receptor 1,FGFR1)、EGFR和KRAS的基因扩增。Li等[31]研究发现MET基因中Y1248H和D1246N突变是MET抑制剂继发性耐药的原因之一。另外,Gimenez-Xavier等[32]从基因组学的角度进行研究发现,2型神经纤维瘤病(neurofibromatosis type 2,NF2)基因对于MET抑制剂的继发性耐药起到关键作用。
总之,MET抑制剂的耐药机制复发多样,既包括了原有MET基因原发性改变,也包括在MET-TKI用药后出现的新突变;既包括其他相关基因的扩增或激活,也包括HGF/MET下游信号通路的改变等(图2)。
4 抑制或逆转MET抑制剂耐药的方法
4.1 不同类型的MET-TKI之间的互换
Ⅰ型MET-TKI 需要与Y1230 堆叠才能结合c-MET,Y1230 突变降低了Ⅰ型MET-TKI 的结合能力。从Ⅰ型MET-TKI转换为Ⅱ型有可能克服此类耐药突变。如1 例MET 阳性晚期肺腺癌患者在应用沃利替尼治疗后进展,发现新的D1228V 点突变,后改为cabozantinib 治疗有效[29]。不同类型的MET-TKI结合位点有所不同,所以耐药机制并不完全相同,因此存在一定的非交叉耐药特点,恰当的二次活检和基因检测可能有助于耐药后选择更为合适的药物。目前认为Ⅰ型MET-TKI 和Ⅱ型MET-TKI 在用药顺序上无明显的优劣,若Ⅰ型MET-TKI 用药后发生耐药,则可以考虑换为Ⅱ型MET-TKI,若Ⅱ型MET-TKI发生耐药,则可以考虑换为Ⅰ型,均可能再次产生疗效。抑制或逆转耐药的机制见图2。
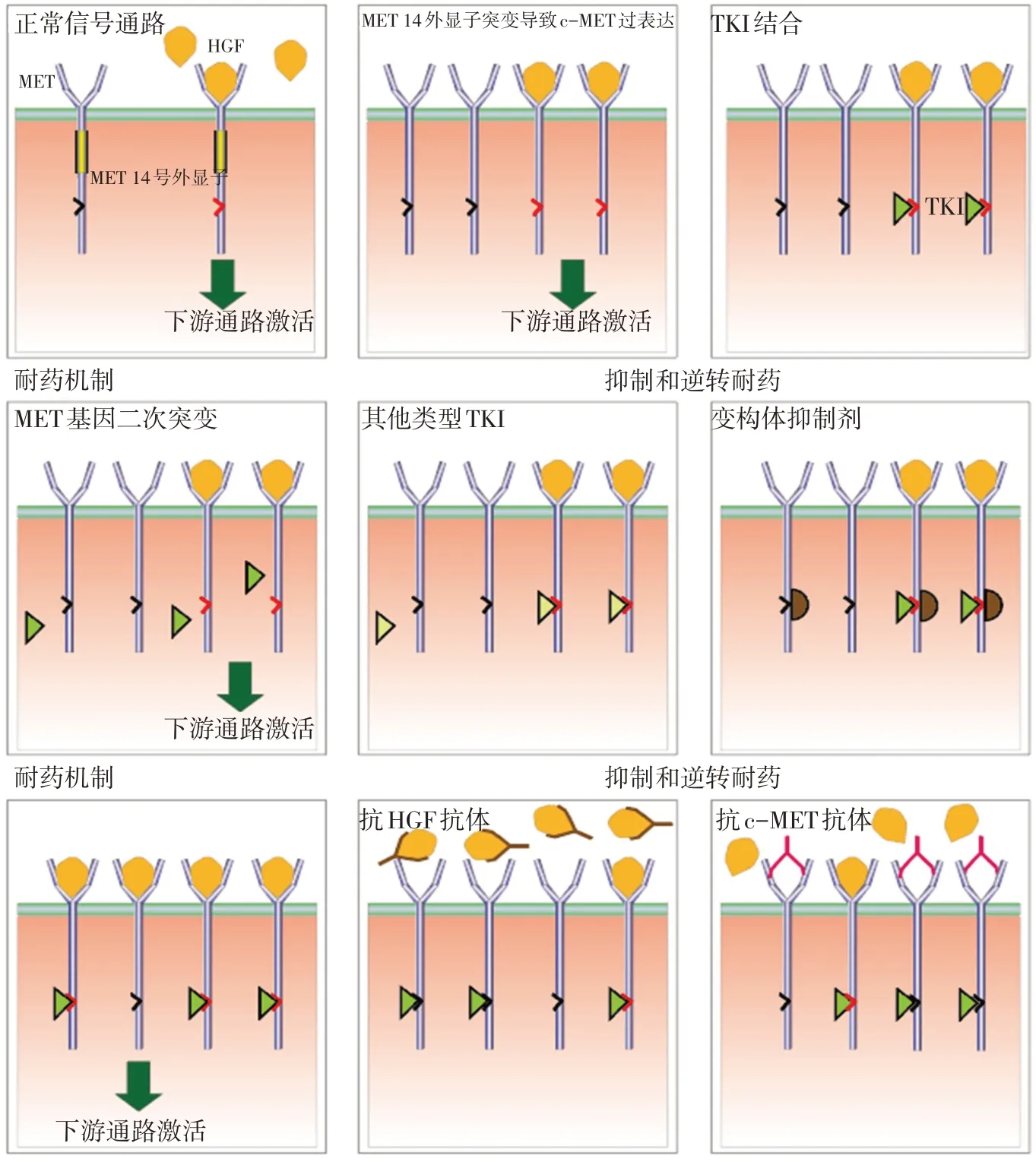
图2 MET抑制剂的耐药机制及其应对策略
4.2 HGF/MET信号通路的多重阻断
在HGF/MET信号通路中,联合应用抗HGF/c-MET单克隆抗体和MET-TKI可能更有效地阻断下游信号通路的激活,从而达到逆转耐药的效果。虽然这一假设在理论上有效,但需要进一步的临床试验验证。
4.3 与免疫治疗的结合
近年来免疫治疗在NSCLC领域取得了突飞猛进的发展。Rivzi 等[33]最先报道肿瘤突变负荷(tumor mutational burden,TMB)与NSCLC患者对帕博丽珠单抗的治疗反应密切相关。MET 14外显子突变的NSCLC患者的平均TMB为6.9突变/Mb(范围0~197.9),低于肺癌总体人群(平均值为10.7突变/Mb),但高于EGFR突变患者(平均值为4.5突变/Mb)和ALK阳性患者(平均值为2.8突变/Mb)[34-35]。EGFR突变和ALK阳性的NSCLC患者,PD-L1表达较低,对抗PD-L1/PD-1药物的治疗效果也较差[36]。虽然MET基因突变患者的PD-L1表达情况尚不明确,但是考虑到TMB的情况,MET抑制剂与免疫治疗的联合有可能克服耐药。国内的一项研究显示,c-MET/PD-1双抗在体外试验中能够有效抑制c-MET和PD-L1均高表达的肺癌细胞的生长、迁移和抗凋亡作用[37]。另外有研究显示,MET抑制剂能够上调肺腺癌PD-L1的表达,HFGF/MET信号通路和免疫逃逸存在一定的关联性[38-39],这也为MET抑制剂和免疫治疗的联合应用提供了理论依据。
4.4 HGF/MET下游信号通路抑制剂
最新的研究发现,PI3K 信号通路的改变可能是MET抑制剂原发性耐药的原因。Jamme等[25]对65例MET 14 外显子突变的晚期NSCLC 患者进行研究,发现其中2 例患者存在PIK3CA 突变,6 例患者存在PTEN 缺失。存在PI3K 信号通路改变的3 例患者在接受MET-TKI治疗后均提示肿瘤进展。另外,METTKI对存在PI3K途径改变的MET 14外显子突变细胞系(包括PTEN 缺失患者来源的细胞系)的增殖无抑制作用。该研究同时提示MET-TKI 与PI3K 抑制剂联合治疗可抑制PI3K 和MAPK 信号转导,并恢复对MET-TKI 的敏感性。因此,为克服MET-TKI 耐药提供了新的治疗思路。
5 结语
MET基因是NSCLC的重要肿瘤驱动基因,METTKI 是治疗NSCLC 患者中具有MET 基因突变人群的有效药物。但是MET-TKI 的耐药不可避免,HGF/MET 信号通路的相关研究,对于抑制和逆转METTKI 的耐药具有重要意义。MET-TKI 与其他药物的联合应用,可能是未来研究的方向。总之,在肺癌的精准治疗时代,分子检测和靶向药物必将为存在HGF/MET信号通路异常的NSCLC患者带来更多的治疗机会和更好的疗效。
