利用大豆四向重组自交系群体定位产量相关性状QTL
2020-06-29王艳殊张佳楠许世超董全中李文滨李文霞宁海龙
王艳殊,田 雨,张佳楠,许世超,董全中,李文滨,李文霞,宁海龙
(1.东北农业大学 农学院,大豆生物学教育部重点实验室,农业部东北大豆生物学与遗传育种重点实验室,黑龙江 哈尔滨 150030;2.黑龙江省农业科学院 克山分院,黑龙江 克山 161606)
大豆(Glycinemax(L.) Merr.)是重要食用植物蛋白质和油脂的主要来源,是我国最重要的经济作物之一,然而国内大豆生产明显不足,截至2000年我国由大豆出口国变为最大进口国,主要原因是产量偏低,因此,提高产量是大豆育种中最重要的目标之一。产量性状是受多个基因控制的数量性状,且易受环境条件的影响,定位大豆产量QTL对于改良产量相关性状具有理论与实践意义。近年来,随着分子遗传学和分子标记技术的迅速发展,构建遗传连锁图谱的分子标记密度逐渐增加,推动了大豆产量相关性状QTL定位的研究。
自1990年Keim等[1]最开始大豆数量性状QTL分析以来,已经在大豆形态与产量等性状方面展开了大量研究。在大豆的产量相关性状中,百粒质量(SW)[2-8]、株高(PH)[9-12]、主茎节数(NN)[13-15]、单株粒数(NS)[16-18]等已有大量的研究和报道。Mansur等[19]利用PI27890 ×PI290136产生的后代重组自交系群体,检测到4个株高QTL,位于连锁群M、J、C2上。Wang等[20]利用IA2008×PI468916 产生的BC后代群体,检测到6个株高QTL,位于连锁群O、K、C2、E、M上,可解释表型变异率在0.74%~0.84%。Guzman等[21]利用Kenwood×LG94-1713产生的BC群体,检测到6个株高QTL,位于连锁群C2、M、J、G上,可解释表型变异率在0.81%~0.92%。Zhang等[12]利用CSSL3228×NN113822产生的F2后代群体,检测到2个株高QTL,位于连锁群F上。Mansur等[22]利用Noir 1×Minsoy 产生的后代重组自交群体,检测到2个单株粒数QTL,位于连锁群C2、M上。Funatsuki等[18]利用Toyomusume×Hayahikan杂交衍生F7后代群体,检测到2个单株粒数QTL,位于连锁群L上,可解释表型变异率为0.83%,Liang等[16]利用BD2×BX10产生的后代重组自交系群体,检测到4个单株粒数QTLs,位于连锁群B1、G、C1上,可解释表型变异率在0.64%~0.79%。Palomeque等[23]利用OAC Millennium ×黑农38产生的后代重组自交群体,检测到3个单株荚数QTL,位于连锁群C2、K上,可解释表型变异率在0.89%~0.96%。Yang等[24]利用Charleston×东农594产生的F2∶14-19后代重组自交群体,检测到1个单株粒数QTL,位于连锁群L上。Li等[17]利用合丰25×Maple Arrow产生的后代重组自交群体,检测到4个单株荚数QTL,位于连锁群D2、I、C2、C1上。Chen等[25]利用Charleston×东农594产生的后代重组自交群体,检测到7个主茎节数QTL,位于连锁群A1、B1、D1a上,可解释表型变异在7.93%~28.46%,Li等[17]利用合丰25×Maple Arrow为亲本杂交得到的F5∶6RIL群体,定位到2个株高QTL,分布在连锁群H上,可解释表型变异率为8.09%,9.82%,4个单株荚数QTL,分布在连锁群D2、I、C2、C1上,可解释表型变异率在6.54%~11.00%,2个主茎节数QTL,分布在连锁群C2、I上,可解释表型变异率为6.79%,17.03%,3个百粒质量QTLs,分布在连锁群M、D2、C1上,可解释表型变异率在7.08%~20.29%。Liu等[9]利用Jinpumkong 2×SS2-2产生的后代重组自交群体,检测到2个主茎节数QTL,位于连锁群D1b、C2上。Hoeck等[26]利用A96-492058×A97-775026产生的后代群体,检测到11个百粒质量QTL,位于连锁群A2、D2、B2、C1、F1、H、L、M上,可解释表型变异在8.6%~28.8%,Kato等[27]利用Ohsuzu×PI595926产生的后代重组自交群体,检测到17个百粒质量QTL,位于连锁群D2、C1、G、K、E、F、O、D1b、I、H、A1上。Hu等[28]利用垦丰1×南农1138-2产生的后代重组自交群体,检测到2个百粒质量QTL,位于连锁群D1b、D1a上。Kulkarni等[29]利用Wiliams 82×PI366121产生的后代重组自交群体,检测到9个百粒质量QTL,位于连锁群D1a、D1b、C2、A2、B2、D2上。
查询SoyBase大豆数据库(https://www.soybase.org/)最新公布的数据发现定位到的大豆产量及相关性状的QTL数量已经超过655个,其中百粒质量229个、株高229个、单株荚数48个、主茎节数37个、单株粒数23个,几乎覆盖所有的连锁群。以往研究中利用2个亲本杂交产生重组自交系,检测产量相关性状的QTL,如亲本遗传差异不大,基因位点无差异,许多QTL不能被检测到。而本试验利用4个亲本杂交产生的后代群体,能克服这个难题。本试验利用四向重组自交系群体多等位基因的优势,探寻有利于产量改良的优异等位基因,为大豆产量性状的改良提供理论依据与技术支撑。
1 材料和方法
1.1 遗传群体设计
用大豆产量相关性状差异较大的4个大豆亲本垦丰14、垦丰15、黑农48和垦丰19,配制双交组合(垦丰14×垦丰15)×(黑农48×垦丰19),F1进行杂交,采用单粒传方法获得RIL群体160个株系的四向重组自交系群体(FW-RIL)。
1.2 田间试验设计
将4个亲本和FW-RIL分别于2013年种植在哈尔滨(E1)和克山(E2)、2014年种植于哈尔滨(E3)、2015年种于克山(E4)和哈尔滨(E5),其中4个亲本各种植一行,田间试验采取随机区组试验设计,小区行长5 m,宽2 m,垄距6 cm,株距5 cm,3行区,3次重复,田间管理同一般大田栽培。
1.3 性状调差
在成熟后,于每个小区随机收获家系10株用于考种,以10株平均值作为小区的代表。调查株高(Plant height,PH)、主茎节数(Main stem node number,NN)、单株荚数(Pod number per plant,NP)、单株粒数(Seed number per plant,NS)、百粒质量 (100-seed weight,SW)等产量相关性。
1.4 遗传连锁图谱的构建
用该FW-RIL群体构建用于数量性状QTL定位的遗传连锁图谱。图谱包含275个SSR引物和20个连锁群,在大豆基因组上覆盖的长度范围是3 636.26 cM,标记间平均长度为15.47 cM。每个连锁群包含遗传标记6~20个,长度在49.36~319.02 cM。
1.5 统计分析
对每个小区10株的数据的平均值进行分析,采用SAS (Statistics Analysis System)9.2的Proc ANOVA对各性状进行方差分析,Proc Varcomp估计的遗传方差、环境方差和基因型与环境互作方差,进而估计广义遗传率。估算公示如下:

基于前文已经构建的遗传图谱[30],应用完备区间作图法(Inclusive Composite Interval Mapping,ICIM)进行QTL定位[31]。首先利用逐步回归的方法在所有标记中筛选重要的标记并估算其效应值,然后利用得出的线性模型调整性状表型值,最后以一维扫描的方法定位 QTL,扫描步长为 1 cM,LOD 值≥2.5。利用软件 GAPL V1.0实现上述运算,得出 QTL定位结果。
2 结果与分析
2.1 描述性分析
对5个环境下各产量相关性状的描述性分析(表1)可看出,FW-RIL的5个产量相关性状在不同环境下都存在较大的变异,各性状存在超亲遗传,说明该群体中各性状有较大分离,该群体对该性状的研究可以有较大的统计推断空间。
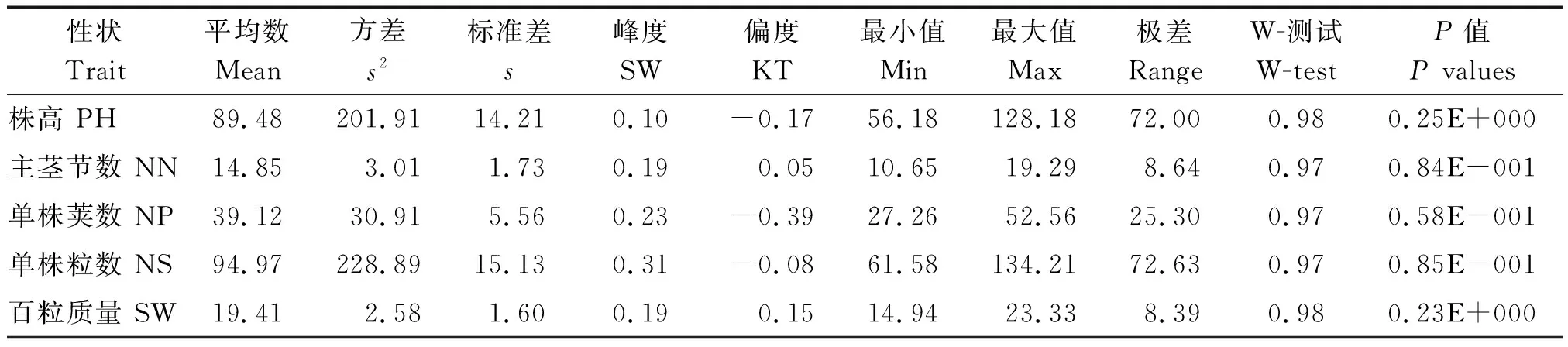
表1 在5个环境下产量相关性状的描述性分析Tab.1 Descript analysis of yield related traits in FW-RIL under five environments
2.2 方差分析和遗传率估计
不同基因型间及环境间效应达到极显著水平(表2),说明群体内这些性状在遗传上有较大差异,并且不同环境下遗传基础亦可能存在较大差异,在不同环境下应用该群体进行基因定位,可能定位到不同的QTL;各性状的遗传力有较大差异,其顺序为株高(0.80)>主茎节数(0.55)>单株粒数(0.42)>百粒质量(0.37)>单株荚数(0.33),表明这些性状的遗传基础不同,受到环境的影响程度不同,检测到QTL的数量也不相同。
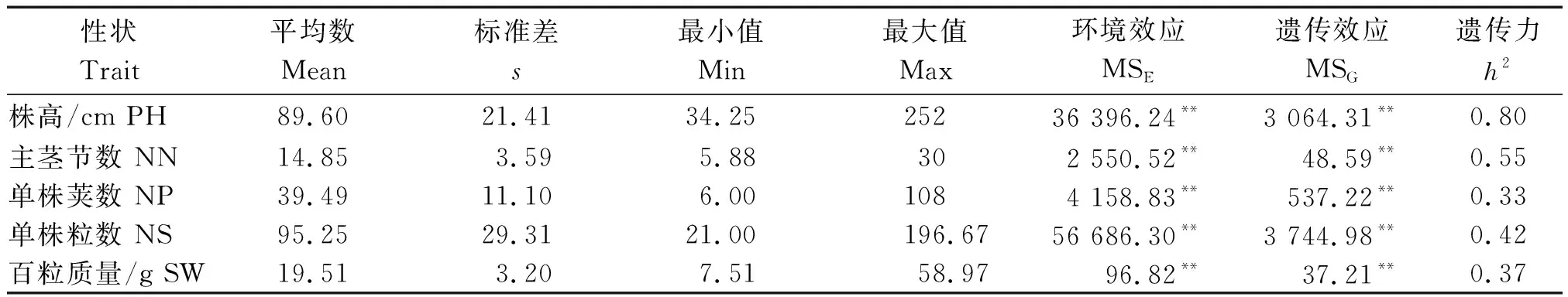
表2 产量相关性状的方差分析和遗传率估计Tab.2 Variance analysis and heritability estimation of morphological and yield-related traits
注:**表示效应在P<0.01水平显著。
Note:**represents the effect significant at the level ofP<0.01.
2.3 QTL的分析
共有28个产量相关性状的QTL在2个以上环境重复检测到(表3、图1),其中10个株高QTL,分布在连锁群B1、B2、C1、C2、D1b、F、G、H、N、O上。qPH-G-2具有最大的可解释表型变异率(Phenotypic variation explained,PVE)和LOD值, 分别为11.72%和8.36,增效基因来自于垦丰14、垦丰15;其他株高QTL为qPH-B1-1、qPH-B2-1、qPH-C1-1、qPH-C2-1、qPH-D1b-3、qPH-F-3、qPH-H-1、qPH-N-1、qPH-O-1。3个主茎节数QTLqNN-G-2、qNN-H-2、qNN-L-1,分别位于连锁群G、H、L上,可解释表型变异率分别为6.55%,5.70%,3.77%,LOD分别为值5.26,3.29,3.23,qNN-G-2、qNN-H-2增效基因来于垦丰14、垦丰15,qNN-L-1增效基因来于垦丰14、垦丰19。9个单株荚数QTLs,分布在连锁群A2、C2、D1a、E、L、M、N、O上,位于连锁群M上的qNP-M-2具有最大的可解释表型变异率和LOD值,分别为11.25%和4.54,增效基因来于垦丰15、垦丰19;其他的单株荚数QTL为qNP-A2-3、qNP-C2-1、qNP-C2-2、qNP-D1a-1、qNP-E-1、qNP-L-1、qNP-N-1、qNP-O-1。2个单株粒数QTLqNS-D2-1和qNS-E-1,分布在连锁群D2、E上,可解释表型变异率分别为8.58%和7.52%,LOD值分别为4.25和4.46。4个百粒质量QTLqSW-A2-3、qSW-D2-3、qSW-F-2、qSW-M-2,分布在连锁群A2、D1、F、M上,可解释表型变异率分别为6.13%,3.83%,5.58%,9.35%,qSW-M-2位于连锁群M上Satt245~Satt677区间内,具有最大的可解释表型变异率9.35%,增效基因来于垦丰14、黑农48,qSW-F-2位于连锁群F上Sat_240~Satt595区间内,具有最大LOD值4.34,增效基因来于垦丰15、黑农48。

表3 5个产量性状QTLTab.3 Five yield-traits for QTLs

图1 定位到的产量相关性状QTL在图谱上的位置Fig.1 Location of yield-related traits QTL on the map
3 讨论
3.1 关于多环境检测QTL
国内外关于产量及产量相关性状QTL的研究日益增多,研究方法也有所不同。以往研究多是单环境下定位QTL,无法确定该QTL定位结果的准确性,基于目前多种QTL定位的方法只是将定位结果简单比较,在多个环境下定位QTL是非常必要, 采用多环境数据分别进行定位,寻找在多环境下重复定位的QTL,互相验证真实性,提高QTL定位的准确性,另外,综合多个环境的分析结果,能增大QTL的检测强度,准确估计QTL的位置和效应。因此,应用多年多点试验进行QTL研究已经成为一种趋势。本研究利用WinQTL Car完备区间作图法(ICIM)对同一研究材料在5个环境下大豆产量相关性状表型数据进行分析,定位到28个产量相关QTLs,这些QTL经过多环境下被重复定位,可认为是真实的。
3.2 关于四向群体
本研究中,试验群体是稳定的重组自交系群体,RIL是通过单粒传法(Single seed descent method,SSD)在F2开始分离单株经多代自交构建而成,大大提高了亲本间遗传多样性的保存,群体内个体基因型纯合,是可以长期使用的永久性分离群体,适合用于图谱的构建和QTL的定位,RIL既可以进行连续性累积,又能方便不同课题间合作。因其永久性群体的特点,还能用来多年多点种植进行基因型和环境互作研究。QTL定位的准确性与群体大小息息相关,群体过小准确性不高,群体过大,准确性提高,但试验工作量太大,费用过高,所以确定合适的群体至关重要,所以本试验选用160个家系作为群体,大小适宜,精确定位到QTL,也减少工作量。本试验不但采用重组自交系群体而且利用四向杂交设计,四向杂交设计群体在每个位点有2个以上的等位基因,与两向杂交设计相比,有3个优势可以提高QTL检测能力。①四向重组自交系群体比双亲本衍生的群体具有更多的多态性遗传标记;②遗传标记具有更高的密度和多态性,增大了统计推断的遗传空间;③连锁群同一位点涉及4个复等位基因,大大提高了QTL检测效率。
本研究在利用前期研究中已经构建的四向重组自交系群体(FW-RIL),用于产量相关性状QTL定位,原始数据来自于3 a 5个环境的田间记录调查和实验室分析,从而定位在多年多点条件下稳定表达的主效QTL位点。可以对定位到的控制产量性状的QTL加以利用来提高大豆的产量。
3.3 不同背景下QTL定位的比较
为验证定位QTL的真实性,将本研究中的定位QTL所在的公共图谱基因组区域与前人研究结果进行比较。本研究共定位到多个环境下稳定检测到的株高QTL 10个,其中,位于连锁群C2上Satt643~Satt363区间内的株高QTLqPH-C2-1在公共图谱上的位置与Mian 等[32]曾报道的株高QTL重叠,位于连锁群D1b上Sat_183~Sat_096区间内的株高QTLqPH-D1b-3在公共图谱上的位置与Sun等[33]曾报道的株高QTL重叠,位于连锁群H上Satt293~Sat_180区间内的株高QTLqPH-H-1在公共图谱上的位置与Lee等[34]曾报道的株高QTL重叠,位于连锁群N上Satt125~Satt62区间内的株高QTLqPH-N-1在公共图谱上的位置与Sun等[33]、Kabelka等[35]、Kim 等[36]曾报道的株高QTL重叠;共定位到单株荚数QTL 9个,其中,位于连锁群A2上Sat_181~Satt228区间内的单株荚数QTLqNP-A2-3在公共图谱上的位置与Zhang等[37]曾报道的单株荚数QTL重叠,位于连锁群C2上Satt291~Satt281区间内单株荚数QTLqNP-C2-1在公共图谱上的位置与Sun等[33]曾报道的单株荚数QTL重叠,位于连锁群D1a上Satt580~AZ302047区间内的单株荚数QTLqNP-D1a-1在公共图谱上的位置与Kuroda等[8]曾报道的单株荚数QTL重叠,位于连锁群L上Sat_405~Satt398区间内的单株荚数QTLqNP-L-1在公共图谱上的位置与Zhang等[37]曾报道的单株荚数QTL重叠,位于连锁群A2上Sct_067~Sat _ 181区间内的百粒质量QTLqSW-A2-3在公共图谱上的位置与Han等[2]、Teng等[38]曾报道的百粒质量QTL重叠,位于连锁群D2上的Satt582~Satt002区间内的百粒质量QTLqSW-D2-3在公共图谱上的位置与Gai等[14]、Zhang等[39]、Li等[17]、Chapman等[40]、Wang等[4]曾报道的百粒质量QTL重叠,位于连锁群F上Sat_240~Satt595区间内的百粒质量QTLqSW-F-2在公共图谱上的位置与Yang等[24]、Mian等[41]曾报道的百粒质量QTL重叠,位于连锁群 M Satt245~Satt677区间内的百粒质量QTLqSW-M-2在公共图谱上的位置与Teng等[38]曾报道的百粒质量QTL重叠。
上述12个产量相关性状QTL位点与国内外报道过的均一致,说明QTL检测准确率较高。利用分子标记遗传图谱,定位控制产量相关性状的QTL,为利用分子标记改良大豆产量潜力提供了有力手段。
3.4 同时控制多种性状的基因组区域
本研究利用5个环境的数据构建遗传连锁图谱,对大豆5个主要产量性状(株高、主茎节数、单株荚数、单株粒数、百粒质量)进行QTL分析,检测到10个株高QTL、3 个主茎节数QTL、9个单株荚数QTL、2个单株粒数QTL、4个百粒质量QTL,但是这些QTL不均匀地分布在连锁群上,一些连锁群上成簇存在,然而其他连锁群包含少量或不包含QTL,大多产量相关性状的QTL集中在染色体的相同区域,即一个基因组区域同时控制多个产量性状,前人也曾报道过,在B1连锁群上的基因组区域Satt426~Satt509内定位到同时控制株高、单株荚数、主茎节数、百粒质量等性状的QTL[25,33,38],Satt197~Satt251内定位到同时控制株高、单株荚数、主茎节数等性状的QTL[25,33,37],Satt251~Satt509内定位到同时控制株高、单株荚数等性状的QTL[25],Satt197~Satt509内定位到同时控制主茎节数、百粒质量等性状的QTL[33],在连锁群C1上的基因组区域Satt565~Satt180内定位到同时控制单株荚数、百粒质量等性状的QTL[17],在连锁群C2上的基因组区域A397_1~A748_1内定位到同时控制株高、主茎节数、单株荚数等性状的QTL[14],在连锁群D2上的基因组区域Satt256~Satt458内定位到同时控制单株荚数、百粒质量等性状的QTL[17],在连锁群H上的基因组区域Satt317~Satt302内定位到同时控制株高、百粒质量等性状的QTL[3],在连锁群L上的基因组区域Sat_286~Satt664内定位到同时控制单株荚数、单株粒数、百粒质量等性状的QTL[8,41],在连锁群M上的基因组区域A343_2~R079_1内定位到同时控制株高、单株粒数等性状的QTL[22,42],本研究共定位到3个控制多个产量相关性状的基因组区域,Satt288~Sct_199同时控制株高、主茎节数等产量性状,Satt204~Satt263和Satt245~Satt677同时控制单株荚数、百粒质量等产量性状。可认为位于这些区间的QTL是多效的。对QTL分布密集的连锁区域进行深入研究,揭示相互间促使遗传信息稳定有序的协调和表达机制,对分子育种具有重要意义。
