成年肺朗格汉斯细胞组织细胞增生症一例报道并文献复习
2020-06-23孙爽赵世峰
孙爽,赵世峰
朗格汉斯细胞组织细胞增生症(Langerhans cell histiocytosis,LCH)是一种罕见的系统性疾病,其特征是CD1a(+)、朗格汉斯细胞组织细胞积聚形成肉芽肿,且会广泛累及器官。朗格汉斯细胞组织细胞起源于髓样树突细胞,肺朗格汉斯细胞组织细胞增生症(PLCH)是成年LCH患者中最常见的一种表现形式,被认为是LCH的一种特殊类型,与吸烟密切相关,其临床表现多种多样,临床极易误诊,目前尚未为成年患者制定明确的治疗方法[1]。虽然PLCH并非在所有病例中均是致命的,但延迟诊断或治疗会导致患者器官功能严重受损、生活质量下降。本文旨在分析1例PLCH患者的临床资料并结合既往文献,以提高临床医师对PLCH的了解,并早期诊治,避免误诊。
1 病例简介
患者,男,25岁,2019-04-30以“咳嗽2月余,气短1月余为主诉”入住中国人民解放军总医院。患者于2019-01-31、2019-02-12、2019-03-07以“咳嗽、胸闷气短”就诊于外院,胸部CT检查示右侧气胸(肺压缩70%),两肺支气管扩张伴感染?;双肺多发气囊肿。外院给予右侧胸腔闭式引流、抗感染、化痰等治疗后症状缓解。患者于外院的电子支气管镜示气管、段以上支气管未有明显异常,为进一步诊治入住本院。既往史:无特殊,否认药物、食物过敏史;个人史:吸烟5年,每天半包;无家族史。入院查体:皮肤无皮疹,浅表淋巴结无肿大,胸廓正常无畸形,胸骨无叩痛;呼吸运动正常,肋间隙正常,语颤轻度减弱;叩诊音清,呼吸规整,双下肺呼吸音粗,未闻及干湿啰音及胸膜摩擦音;心脏、腹部查体正常,无肝脾大,无关节骨痛,无神经肌肉异常,无下肢水肿。血常规检查:白细胞计数12×109/L〔参考范围:(4~10)×109/L〕、中性粒细胞分数0.82(参考范围:0.50~0.70)、淋巴细胞分数0.09(参考范围:0.20~0.40)、血小板计数303×109/L〔参考范围:(100~300)×109/L〕、白介素6 40.1 ng/L(参考范围:56.4~150.3 ng/L)、红细胞沉降率41 mm/1 h(参考范围:男性:0~15 mm/1 h,女性:0~20 mm/1 h),C反应蛋白(CRP)、红细胞计数等均在参考范围。免疫炎性指标:免疫球蛋白E(IgE)284 U/ml(参考范围:0~100 U/ml)、免疫球蛋白G(IgG)1 650 g/L(参考范围:700~1 660 g/L)、r球蛋白19.5%(参考范围:11.1%~18.8%),余免疫指标均在参考范围。生化、凝血检查均未有明显异常,淋巴细胞亚群未有异常,风湿免疫指标未有阳性结果,甲状腺、性腺激素轴均在参考范围,肿瘤标志物均正常。患者入院时血气分析(未吸氧):pH值7.3、动脉血二氧化碳分压(PaCO2)44.9 mm Hg(1 mm Hg=0.133 kPa)、动脉血氧分压(PaO2)62.1 mm Hg、动脉血氧饱和度(SaO2)90.6%、乳酸(Lac)1.6 mmol/L、氧合指数296 mm Hg;肺功能:第1秒用力呼气容积占用力肺活量百分比(FEV1/FVC)75.5%,第1秒用力呼气容积占预计值百分比(FEV1%)78.5%,最大通气量占预计值百分比(MVV%)88.9%,用力呼气中期流速占预计值百分比(MMEF)50.7%。意见:小气道通气功能障碍,气道阻力及肺周边弹性阻力增高。肺弥散检测:肺一氧化氮弥散量正常,肺总量残气容积正常,残总百分比正常,呼出气一氧化氮(FeNO)12 ppb。入院肺CT检查结果示两肺多发囊性病变伴结节及空洞形成(见图1),多考虑LCH可能。入院后患者炎性指标轻度升高,给予头孢曲松、左氧氟沙星抗感染治疗后炎性指标正常,两周后再次复查胸部CT结果示较前未有改善(见图2),排除感染因素引起肺囊泡改变。后行外科胸腔镜肺活检术,切取5 cm×2.5 cm×1 cm病变肺组织,表面见泡状物2.1 cm×1.8 cm×1 cm(见图3),病理结果诊断大部分肺泡扩张,部分肺泡间隔断裂融合,免疫组化结果示上皮样细胞中CD1a(+)、CD68(+)、S-100(+)、CD21(-)、CD35(-)、抗黑素瘤特异性单抗(HMB45)(-)、T细胞识别的黑色素瘤抗原(Melan-A)(-)、平滑肌肌动蛋白(SMA)(-)、CD34(血管壁+)、细胞角蛋白(CK)(-);符合LCH。正电子发射计算机断层显像扫描(PET/CT)检查结果示双肺部多囊性病变及结节,余躯干部位未有转移,无其余系统累及;心脏超声检查结果示肺动脉压在参考范围,未有异常结构变化,行组织及血液LCH基因检测及BRAPV600E、MAPK2K1基因检测,结果显示,血液基因组EPHA5基因错译突变,外显子1,cDNA 173G>C,氨基酸改变Ser58Thr,丰度0.75%;组织及血液均未检测到BRAPV600E、MAPK2K1基因突变。予以严格戒烟,确诊后予以甲泼尼龙48 mg口服,1次/d,1月后复查肺部CT(见图4)及肺功能,较前有所好转。
2 讨论

图1 入院时肺CT检查结果Figure 1 Lung CT result on admission
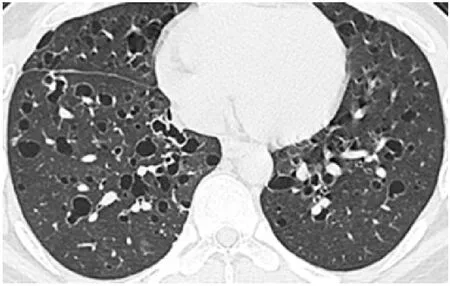
图2 入院两周后肺CT检查结果Figure 2 Lung CT result two weeks after admission

图3 病变的肺组织Figure 3 Lesion lung tissues

图4 入院1月后复查肺CT检查Figure 4 Reexamination of lung CT 1 month after admission
PLCH是一种少见的弥漫性间质性肺疾病,可影响单个或多个器官,一般好发于年轻吸烟者。目前PLCH的发病率尚不清楚,但其占成年人弥漫性间质性肺病的3%~5%[2]。因PLCH较为罕见,且对男女均有同样影响,但男性患者在较早的年龄中就会出现症状[3]。本文就本院收治的1例PLCH患者临床资料并结合既往文献分析PLCH的临床特征、诊断及治疗。
2.1 临床特征及诊断 PLCH最常见的症状是干咳、呼吸困难和胸痛;非特异性症状如不适、体质量减轻、盗汗和发热。有研究表明,约20%的患者在疾病早期无症状,而有一半患者出现与气胸有关症状[4]。本例患者为年轻男性,具有吸烟史,去藏区后出现咳嗽、胸闷、呼吸困难,反复气胸起病,偶有干咳;肺功能检查示轻度阻塞性通气功能障碍,残气量、肺弥散、舒张试验及FeNO均正常。有研究表明,50%以上PLCH患者的肺功能存在阻塞性通气功能障碍,而限制性通气障碍或肺功能正常的患者所占比例较小,气流限制的严重程度与肺实质受累程度有关[5]。肺活量和总肺活量降低者较少,在复发性气胸和胸膜切开者可出现肺活量降低。PLCH最常见的异常是肺对一氧化碳的弥散能力降低,70%~90%的患者可观察到[6]。本例患者肺功能轻度异常改变,考虑与患者年龄及体质因素有关,但仍需对患者肺功能变化进行随访。PLCH经高分辨率CT初步表现为直径1~10 mm的支气管周围和小叶中心结节,边界不规则,随病情进展呈囊状表现,结节和囊肿常同时出现,不同大小及不规则形状的囊肿在疾病后期以合并为主,随着疾病的发展,囊肿最初的厚壁变薄[1]。有广泛的肿瘤性炎症和传染病具有相似的放射学表现,扩大了鉴别诊断的范围,包括肺淋巴管平滑肌瘤病(好发于女性且存在性腺激素分泌异常)、支气管扩张、先天性肺囊肿、肺结核、血管炎、转移性结肠癌、结节病、过敏性肺炎和感染[7-9],易误诊及漏诊。本例患者肺部高分辨率CT表现为囊性病变,并倾向于合并;在同一或不同的CT检查结果上可见混合空洞和孤立结节,双侧多发结节主要位于上、中区,囊腔多位于中下肺。影像学变化结合症状及体征并完善肿瘤标志物、性腺轴、结核、风湿免疫等指标筛查后,考虑为PLCH可能性较大,其为多器官受累疾病,10%~20%的成年LCH患者表现为肺外受累,如尿崩症(DI)、内分泌、皮肤和骨病。本例患者PET/CT检查结果示未有其他器官及组织受累,为单纯肺受累,予以行外科胸腔镜肺活检术切除右侧肺叶部分病变组织,免疫组化结果示朗格汉斯上皮样细胞CD1a(+)、CD68(+)、S-100(+)、CD21(-)、CD35(-)、HMB45(-)、Melan-A(-)、SMA(-)、CD34(血管壁+)、CK(-),符合LCH。因此病理组织细胞检查是确诊LCH的重要依据。
PLCH的初步诊断主要依赖于临床表现、胸部CT结果示囊肿和结节,并主要可见于上肺和中段,发现可疑病灶进行组织学确认。PLCH明确诊断主要取决于临床表现和在电子显微镜下可见的活检材料中朗格汉斯细胞的鉴定[10]。在疾病的早期阶段,电子显微镜检查显示主要是位于支气管周围间隙形成不良的炎性结节,后炎性浸润延伸到肺泡间隙,使肺结构变模糊,结节含有多种炎性细胞,其中T细胞、巨噬细胞、单核细胞和朗格汉斯细胞相对较多,而电子显微镜观察显示,在细胞质中有所谓的Birbeck颗粒,具有LCH的病理特征,且在免疫组织化学检测中也可发现抗CD207、S蛋白和CD1a抗原的表达,对诊断有重要意义。在疾病晚期,结节的中央部分形成腔隙,组织细胞性肉芽肿可能伴随肺损伤,朗格汉斯细胞较少,以囊性和纤维型为主[11]。据报道,25%~65%的PLCH患者与丝裂原活化蛋白激酶(MAPK)下游BRAFV600E、MAPK2K1基因突变相关,RAF蛋白协调与细胞增殖、分化、存活和凋亡相关的信号通路[12]。因此BRAF基因突变被认为有助于克隆细胞增殖。本例患者行组织及血液LCH基因BRAPV600E、MAPK2K1检测,血液基因组结果示EPHA5基因错译突变,未有BRAPV600E、MAPK2K1基因突变。有研究报道,BRAFV600E在多系统的LCH患者中比在孤立性疾病患者中更常见[13]。
2.2 治疗 本例患者确诊后,考虑为单系统受累,采取严格戒烟,同时给予激素治疗后症状有所缓解。研究表明,LCH的治疗取决于疾病的传播、受影响的器官及其损伤程度,在单纯性PLCH患者戒烟的情况下,约50%患者可根据临床客观指标(包括肺功能、影像学、6 min步行试验等)评估病情[10,14-15]。有研究表明,当患者出现强烈的呼吸系统症状,特别是肺结节性病变,研究者建议采用系统性类固醇治疗,建议每月1 mg/kg泼尼松,并逐渐减少剂量,治疗时间不超过6个月,但这种疗法还未被临床验证[4]。对于多系统受累患者无论是否累及关键器官、单系统受累合并多个病灶、单系统受累合并特定部位病变患者均推荐全身化疗。但目前还未有治疗标准。虽然长春花碱和泼尼松对儿童的化疗是有效的,但在成年人的治疗效果不太理想[16],因此建议对成年LCH患者可采取细胞还原疗法(甲氨蝶呤、长春新碱、6-巯基嘌呤、依托泊苷)[17-18]。有BRAF突变的耐药患者,推荐使用MAPK通路的药物,但需进一步评估其有效性[13]。本例患者服用激素后逐渐减量,1个月后随访肺功能、影像学检查有所好转,考虑目前治疗有效;在激素减量治疗过程中,还需密切观察患者病情并注意对患者并发症的管理,尤其是前毛细血管肺动脉高压[16]。
总之,PLCH是一种罕见的疾病,好发于吸烟的年轻男性,CT检查结果示有自发性气胸的囊性肺病,应考虑该病。LCH诊断主要依赖于病理学及免疫组化等;其为多系统受累疾病,诊断后需行全身多系统检查是否累及其他器官,以便制定治疗方案及对症支持。LCH会引起阻塞及限制性功能障碍,需与其他疾病相鉴别,该疾病主要影响扩散能力,并可引起肺动脉高压、心肺功能异常,因此应严格定期复查肺功能、心脏超声,并予以及时治疗。戒烟对治疗PLCH至关重要,如果激素无效可应用化疗及细胞抑制剂,必要可应用基因靶向治疗。
作者贡献:孙爽进行试验设计与实施、资料收集整理、撰写论文并对文章负责;赵世峰进行质量控制及审校。
本文无利益冲突。
罕见病解读:
目前肺朗格汉斯组织细胞增生症(PLCH)的流行病学情况尚不清楚。在成年人中,PLCH主要发生在年轻的吸烟者或既往吸烟者(90%的病例中),20~40岁是疾病的高发年龄,且其他吸入性暴露因素可能与疾病也有关。朗格汉斯细胞组织细胞增生症(LCH)目前被认为是一种具有多种临床表现的炎性骨髓增生。吸烟在肺部病灶的发展中是重要的触发因素。其他的外源性因素,如病毒感染,虽然怀疑与疾病相关但尚未被确认。
该病的自然病程是难以预料的,疾病可自然缓解,因此对于PLCH患者管理最重要的是密切随访。PLCH的自然病程及预后目前缺乏明确数据。对所有患者来说戒烟是最重要的。一项纳入102例经活检证实为PLCH的回顾性研究表明,PLCH的中位生存期为12.5年。部分患者戒烟后可不经治疗完全缓解。多达40%的患者在确诊后的2年内可能出现第1秒用力呼吸容积(FEV1)或一氧化碳弥散量(DLCO)降低超过15%。5年内,约50%的患者将会随着时间的推移出现肺功能受损和阻塞性肺疾病。在部分患者中,由于吸烟引起的慢性阻塞性肺疾病,肺功能会持续恶化。在10%~20%的患者中,诊断初期即存在严重的功能损害,随后出现进行性呼吸衰竭及慢性肺心病。目前只有肺动脉高压是患者预后不良的明确预测指标。研究发现持续吸烟会增加肺功能恶化的风险。
