多孔炭基二氧化碳电催化材料研究进展
2020-06-22董灵玉葛睿原亚飞唐宋元郝广平陆安慧
董灵玉,葛睿,原亚飞,唐宋元,郝广平,陆安慧
(大连理工大学化工学院,精细化工国家重点实验室,辽宁省低碳资源高值化利用重点实验室,辽宁大连116024)
引 言
二氧化碳(CO2)是人类活动导致的主要温室气体。虽然生物碳循环(光合作用)、工业过程(尿素生产、干冰制造、石油开采等)等利用了一部分CO2,但是现代社会的高速发展导致CO2排放与消耗不对称问题日益突出,大气中CO2浓度逐年增高[1]。通过催化过程将CO2加氢还原为高附加值化学品是研究人员寻求的目标。CO2作为线性对称分子,碳原子的两个sp 杂化轨道分别与两个氧原子sp2杂化轨道生成两个σ 键,碳原子的两个p 轨道分别与两个氧原子的p轨道形成π键[图1(a)],CO2离域π轨道上电子形成两个稳定的三中心四电子的大π 键(即π43键),其中电子云主要束缚在氧原子上,碳原子则为缺电子中心[图1(b)]。CO2的分子结构决定了其弱电子给予体及较强电子受体性质及其稳定性(ΔG = 394.4 kJ·mol-1),因此要实现CO2活化,通常需要强电子供体,有效催化剂及高能量输入[2]。常见的CO2活化过程主要包括:(1) 生物活化(光合作用暗反应);(2) 化学活化(加氢催化);(3)电化学活化(质子电子耦合);(4)光化学辐射活化等[图1(c)]。
CO2的生物活化时时刻刻都在发生,维持着总体碳循环;化学活化在化工生产中发挥重要作用,涉及CO2的主要催化反应包括CO2加氢制甲醇或乙醇、CO2与甲烷制合成气、CO2制乙酸和CO2制有机精细化工产品等[3]。目前,绿色可持续能源是社会发展的共识,其中基于可持续能源(太阳能、风能、地热、潮汐能SWGH)的电能是最重要的代表,电动社会也是未来发展的趋势。在此背景下,电催化还原CO2引起广泛关注[4]。电催化还原CO2研究主要集中于催化剂开发、电极集成工艺以及电解体系构建等方面,其中催化剂开发是第一步。因此,开发高效、稳定、具有高CO2还原活性的电催化材料成为研究的热点。CO2还原反应是一个多质子耦合、多电子转移的过程,最终产物包括一氧化碳(CO)、甲酸、甲醛、甲烷、甲醇、C2碳氢化合物和含氧化合物。CO2电催化材料主要包括以下几类:(1) 金属(Cu、Au、Ag、Pd等)[5-6];(2)金属氧化物(SnO2,Cu2O,Co3O4等)[7-8];(3)共价有机骨架[9-10]与金属有机骨架[11];(4)分子催化剂[12-13];(5)非贵金属多孔炭基催化材料[14-16]。炭基催化材料不仅具有导电性好的天然优势,还有孔结构可控、比表面积大、稳定性突出等结构特色;在CO2电催化中取得一系列重要突破[17-18]。本文综述近年来多孔炭基CO2电催化材料取得的重要研究进展。首先讨论炭基CO2电催化材料的核心参数(孔结构、表面化学、形貌)的协同调控策略;随后以典型的多孔炭基催化材料为例分析其应用于电催化CO2还原的活性增强策略;最后对多孔炭基催化材料的研究进展进行总结,并对电催化还原CO2的前景与挑战进行展望。
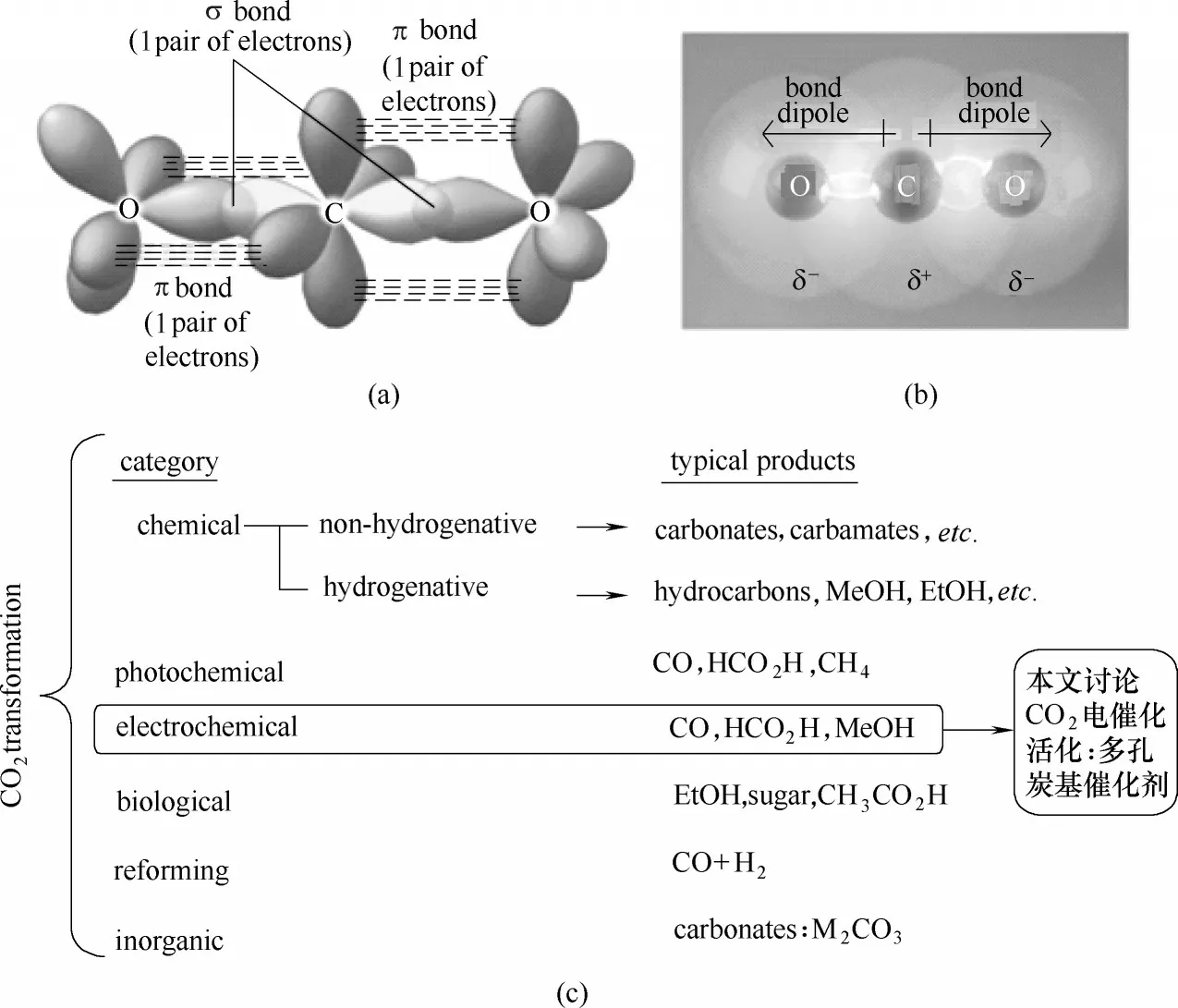
图1 CO2分子杂化轨道及域电子分配(a),CO2原子电负性(b)和CO2活化方式(c)Fig.1 Hybrid orbital of CO2and delocalized electrons(a),C and O element’s electronegativity in CO2(b)and the methods to activate CO2(c)
1 炭基CO2 电催化材料的结构调控策略
碳元素具有sp、sp2、sp3三种主要的轨道杂化方式,因此成键方式灵活[19]。主要基于sp2/sp3杂化形式成键的炭材料由于具有优良的导电导热性能、丰富的孔隙等性质,广泛应用于储能、吸附、催化等领域[20-25]。按材料结构,炭基材料分为结晶态及非晶态。结晶态的炭材料主要包括石墨、富勒烯、碳纳米管、石墨烯[26];非晶态炭材料包括炭黑、活性炭、微介孔炭分子筛。虽然炭材料本身导电性优异,但是其自身无电催化CO2活性,因此要对其进行功能化与结构调控。炭材料的结构调控主要从孔结构、表面化学及形貌三个方面考虑。
1.1 孔结构调控
关于孔径大小,1985 年国际纯粹与应用化学协会(IUPAC)对其进行了规定,分为微孔(micropore)——孔径小于2 nm 的孔、介孔(mesopore)——孔径介于2~50 nm 的孔、大孔(macropore)——孔径大于50 nm的孔[27]。2015 年IUPAC 又进一步规定,微孔又分为孔径小于0.7 nm 的超微孔(ultramicropore)和孔径大于0.7 nm的极微孔(supermicropore)[28]。
1.1.1 微孔炭材料 多孔炭多为微孔材料,其直径契合小分子的尺度,适合用来吸附气体小分子。例如CO2的动力学直径约为3.3 Å (1 Å=0.1 nm),孔径为6~7 Å 的微孔对其具有较高吸附势能,表现出对CO2的高选择吸附。另外,丰富的微孔可以促进催化剂活性组分的负载和分散,有利于催化反应活性的提高。对于多孔炭微孔的调控已有大量研究。例如,分子筛硬模板法[29-30]、碳化物氯气刻蚀法等[31]可以实现对微孔孔径精确调变。Ryoo课题组[32]利用稀土La元素的d轨道与碳源(乙烯、乙炔等)之间d-π相互作用,通过稀土镧离子(La3+)改性后的LaY 分子筛为模板,促进乙烯优先在分子筛孔道内沉积成碳,制备了具有三维石墨烯结构的炭分子筛。Oschatz 等[33]通过对聚碳硅烷气凝胶热解、氯化,制备了一种具有可调孔隙的炭气凝胶,比表面积和微孔体积可达2410 m2·g-1和0.63 cm3·g-1;他们发现降低热解温度有助于更多微孔的形成。Shao等[34]以八苯基环四硅氧烷为底物,以氰尿酰氯为交联剂,制备有机-无机交联聚合物作为前体制备多孔炭材料,比表面积、微孔体积及孔隙率的调变范围分别为237~2058m2·g-1、0.10~0.93cm3·g-1及79.4%~88.5%。1.1.2 介孔炭材料 相比于微孔,介孔有利于CO2分子在孔内的扩散,传质效率更高。通过嵌段共聚物软模板、介孔固体硬模板可以制备有序介孔炭[35-36],进而可以分析介孔孔径、孔型对于CO2电催化的影响。例如,Hursan 等[37]以不同粒径的SiO2纳米球作为硬模板,以邻苯二胺为碳源,制备了介孔炭基CO2电催化材料,研究了形貌-活性-稳定性规律。作者发现介孔炭的形貌、润湿特性和曲率效应共同导致CO2电催化行为在活性和选择性上的显著差异,证实介孔的存在有利于提高CO 选择性和电流密度。
1.1.3 多级孔炭材料 大孔-介孔-微孔串联多级孔结构进一步促进了CO2的吸附-扩散动力学,对于CO2电催化研究也有启示作用。例如,Deng 等[38]结合嵌段共聚物和二氧化硅软-硬模板法,制备了具有介孔-大孔的多级孔炭材料。Wang 等[39]报道了一种多级孔结构氮掺杂炭膜,发现大孔可以提供CO2快速扩散的通道,微孔和介孔贡献了主要比表面积和活性位点,提高了电催化还原CO2的活性。
1.2 表面化学
多孔炭材料的表面化学的调控也是提高其性能的重要手段。CO2分子电四极矩不为零,可视为电四极子;因此强极性表面可增强对CO2的诱导,促进其吸附及活化。多孔炭材料的表面化学调控方法包括改变其极性、引入缺陷结构、掺杂杂原子等,这通常也是多孔炭催化材料活性的来源。
1.2.1 表面极性 CO2的电催化还原反应大多数情况下在水系电解液进行,水合CO2向活性中心迁移的速率对还原反应的发生起到至关重要的作用。因此,增加浸润性也是增强电催化还原反应的重要手段。由于绝大多数炭材料自身显示疏水性,在水溶液体系中完成电催化还原反应存在先天弱势,对其进行亲水改性成为引起关注的研究方向。Hao等[40]对一种块状多孔炭的表面进行亲水改性,加强了其表面异质性与微孔结构之间的协同效应,使之表面具有良好的润湿性,其后又通过氮掺杂[41]和硼-氮双掺杂[42]制备了具有丰富微孔结构和高表面润湿性的炭材料,在电催化还原反应中表现优异性能。但是,炭材料的亲水性对电催化还原反应并非单一的促进作用,也有可能不利于水系环境中气体产物的及时逸出,影响反应的连续进行,因此需要综合评估亲水疏气或亲气疏水的影响[43-44]。
1.2.2 缺陷结构 多孔炭材料的常见缺陷结构包括空位、位错、边缘等。通常缺陷结构的引入会改变材料极性,此外,这些缺陷也赋予其特殊的性能,引入缺陷或者对缺陷进行调控是多孔炭材料的重要研究方向[45-49]。在电催化还原CO2中,催化材料的缺陷会带来较多的活性位,通过对碳纳米管、碳纳米笼等材料的研究[50-52]以及结合DFT计算[53],证明了边缘缺陷位比基面具有更高的催化活性。因此,调控炭材料的缺陷是提高电催化反应活性的重要途经。例如,Joo 等[54]研究发现具有丰富边缘缺陷的石墨化介孔炭的电催化活性是具有丰富基面的碳纳米管的28 倍。Hu 等[55-56]研究了纳米炭笼材料的本征缺陷对于增强电催化活性的影响。最近,Cohen-Karni 等[57]研究表明边缘位点丰富的三维石墨烯表现出显著提升的活性,高选择性,且催化活性与石墨烯边缘位点密度呈正比例关系。三维石墨烯的边缘缺陷位点通常表现出与面内不同的电荷分布,进而改变CO2化学吸附强度,降低了电子质子耦合形成*COOH 中间体的活化能,提高了CO2的活性与选择性。通常引入边缘缺陷的途径包括:化学氧化法,机械球磨法,等离子体刻蚀法等[58]。这些工作都为设计高效CO2电催化材料提供了指导。
1.2.3 杂原子掺杂 在多孔炭材料中引入杂原子来实现特定的性能,不同原子的引入导致材料的性能各不相同。严格讲,杂原子掺杂也是引入缺陷的一种形式[59]。比如,氮原子掺杂研究最为广泛,其引入对导电性、表面极性、金属原子调节配位环境等带来一系列影响[60-61]。杂原子的引入对多孔炭材料催化活性具有明显增强,尤其非金属杂原子与过渡金属原子共掺杂对于提高CO2电流密度具有显著促进作用,这一点将在后续章节详加叙述。
1.3 形貌
随着炭材料结构控制的精细化程度日益提高,研究人员发现炭基催化材料的活性不仅取决于其化学组成, 还与它们的形貌特征,例如维度、形貌、粗糙度等直接相关。这对多孔炭基电催化材料应用于CO2电催化同样适用。通过筛选前体、调控反应条件、创新炭化过程可以实现新结构多孔炭的创制,作为模型材料在研究CO2电催化构效关系中发挥重要作用。碳原子具有独特成键能力,可通过不同的杂化轨道(如sp,sp2,sp3)成键,这为其形貌结构灵活调变提供可能。根据碳纳米结构的维度可分为以下四类。(1)零维纳米结构:富勒烯,碳量子点,炭球。(2) 一维纳米结构:碳纳米管,炭纳米纤维。(3)二维纳米结构:炭膜。(4)三维多孔炭。接下来,本文以多孔炭球、碳纳米管、炭纤维、多孔炭膜以及整体式多孔炭的调控为例进行论述。
1.3.1 多孔炭球 多孔炭球由于具有大的比表面积,低密度和高负载能力,在微纳米反应器、催化、能量存储等诸多方面得到了应用。在电催化方面,多孔炭球也是优良的催化剂载体,孔道的存在意味着电解质可渗透到炭球中,到达孔道内表面的活性位点[62]。此外,多孔炭球还可改善催化剂的导电性。Jesica等[63]采用溶胶-凝胶法制备了镍负载的介孔炭纳米球,增加了镍活性位点与电解液的接触,促进了电催化CO2还原。Chen 等[64]以二氧化硅为模板,合成了铁氮共掺杂介孔炭纳米球(Fe-N-PC),Fe-NPC 具有高分散的活性位点,可将CO2转化为CO,在-0.49 V 表现出90%的法拉第效率和11.44 mA·cm-2电流密度,展现出了优异的电催化还原CO2性能。
现阶段,单分散炭纳米球的合成仍面临挑战。通过引入新型苯并嗪聚合体系,Wang 等[65]建立了一种具有精确控制尺寸和高分散性的炭纳米球的合成方法,推动了单分散炭纳米球的发展。该体系使用间苯二酚、甲醛和1,6-己二胺三组分共聚,以F127 作为表面活性剂,在精确控制反应温度下合成了可控制尺寸的纳米球,球径可从105~250 nm 连续调控。
1.3.2 多孔碳纳米管、炭纳米纤维 杂原子掺杂型碳纳米管、炭纳米纤维可直接用作CO2催化材料,Wu 等[66]利用液相化学气相沉积法合成了N 掺杂的碳纳米管,然后将碳纳米管经聚乙烯亚胺改性后,可将二氧化碳直接还原成丁烷。但在现阶段,碳纳米管、炭纳米纤维由于其自支撑结构和孔结构有利于活性位点在径向的暴露,且其具有良好导电性,主要是被用作催化剂载体。Yang 等[67]合成了均匀分布的Cu单原子炭纳米纤维(CuSAs/TCNF)。如图2所示,CuSAs/TCNFs 产生了大量有效的Cu 单原子,用作CO2还原反应的催化材料,在液相中唯一产物为甲醇,法拉第效率为44%。Zhang 等[68]采用静电纺丝结合热解的方法合成了由N掺杂炭纳米纤维支撑的CoxNi1-x纳米合金,用于CO2还原的电催化材料,实现了高效CO2还原反应。Zhou 等[69]将合成的花生状碳纳米管(FP5N)和掺N 的FP5N 进行CO2电催化还原,发现其性能已经与过渡金属催化剂相当。

图2 CuSAs/TCNF结构表征Fig.2 Structural characterization of CuSAs/TCNF
目前,化学气相沉积和电纺丝技术被广泛用于多孔炭纳米纤维的制备。在Modi 等[70]的研究中,利用催化化学气相沉积法制备了含N的多尺度炭纳米纤维。但是,开发一种用于合成具有高表面积,大孔径和丰富表面功能的炭纳米纤维的简便方法仍然是一项巨大的挑战。
1.3.3 多孔炭膜 多孔炭膜,例如石墨烯膜,具有高比表面积、优异的导电导热性,被广泛应用于分离、电化学储能和催化剂载体等。当杂原子掺杂功能化后,可以有效地诱导掺杂剂附近C 原子上的电荷和自旋密度发生变化,改变其电子性质并产生催化活性中心,在电催化CO2还原中表现出优良的性能,有些已经接近甚至优于贵金属催化剂[71]。Mao等[72]将Si 原子掺杂到石墨烯边缘(Si@G)上,Si@G 在电催化CO2还原过程中表现出了非常高的选择性。杂原子掺杂石墨烯作为一种很有前景的CO2还原的非金属催化剂,为进一步提高CO2转化效率开辟了新途径。而石墨烯由于具有明确的原子结构和多种化学配位环境,近年来,单原子电催化催化剂载体研究也引起了人们广泛兴趣。Zhang 等[73]通过在石墨烯上对血红素和三聚氰胺分子进行充分热解,得到了铁单原子电催化剂。由于负载在石墨烯上的高分散FeN5活性位点的存在,该催化剂在0.35 V的低过电势下表现出高达97.0%的法拉第效率,胜过所有基于Fe-N-C 的催化剂。同样地,Jiang 等[74]将镍单原子负载在石墨烯纳米片,得到镍单原子催化剂。
目前,多孔炭膜的合成主要是通过硬模板和软模板,有机聚合物前体热解,化学和物理气相沉积。如Gierszal等[75]报道了一种具有大孔体积,均匀孔径和受控厚度的均匀炭膜的合成方法:首先在二氧化硅晶体或二氧化硅胶体聚集体的孔壁上形成均匀的聚合物膜,然后进行炭化和模板溶解,即可得到炭膜产物。

图3 整体炭宏观形态与电子显微镜照片Fig.3 Macroscopic morphology and electron microscopy images of typical carbon monolith
1.3.4 三维结构多孔炭 整体式多孔炭通常具有几个明显的优势,如低压降、快速传热和传质、高接触效率、易于操控等。整体结构炭的合成通常有溶胶-凝胶法、纳米浇铸法和自组装方法等[76]。近年来,通过共聚物分子模板与碳前体的自组装,直接合成整体多孔炭材料,取得了很大的进展。Lu 课题组[77]基于苯并嗪化学体系,合成了一种新型的多孔整体炭,展现出了双连续多级孔结构、含氮骨架和高机械强度(图3)。该整体炭即使在水汽存在的情况下,也能表现出优异的CO2吸附和分离能力,并且容易再生。这为整体式多孔炭应用于CO2电催化气体扩散电极材料提供了可能。
2 多孔炭基二氧化碳电催化材料
多孔炭基催化材料最早应用于CO2电化学还原反应借鉴了其在氧还原反应应用的经验[78-79]。与氧还原反应类似,多孔炭材料本身没有电催化CO2还原活性。因此,活性的引入首先从非金属杂原子掺杂开始,研究人员报道了一系列非碳杂原子掺杂多孔炭基催化剂;随后,研究发现过渡金属与氮原子共掺杂可以显著提高CO2还原活性;同时研究人员还探索了将均相催化剂与非均相催化剂融合的方式,开发了多孔炭负载的CO2还原活性的配合物催化剂。下面将结合典型的研究进展实例,对这几类CO2电催化剂进行讨论。
2.1 非金属杂原子掺杂/炭电催化材料
金属催化剂已被研究用于CO2还原,但在成本、选择性、析氢竞争反应控制方面都有待改善[80]。近年来,炭基催化剂显示出高的催化活性、耐久性和选择性,是一种有前途的电催化剂[81]。首先,与贵金属催化剂相比,炭材料具有可调变的多孔结构、高表面积、耐酸碱性、耐高温稳定性、环境友好性[82]。此外,多孔炭材料具有高的孔隙率,有利于CO2的吸附和电催化还原CO2时电解质的快速渗透[83]。
鉴于中性碳原子不具有活化CO2分子的能力,研究人员采用杂原子掺杂的办法,在各种多孔炭材料(如炭纤维、碳纳米管、石墨烯、金刚石、纳米孔炭和石墨烯点)上掺杂N、S 和B 等,制备CO2还原催化剂[84]。掺杂杂原子的碳纳米材料的制备方法主要包括含有杂原子的前体原位掺杂及后处理掺杂(如氨气活化)。原位掺杂可以均匀地将杂原子结合到整个结构中,而后处理掺杂杂原子往往会导致表面功能化,而不改变整体性质[85]。图4(a)示意了杂原子B、N、P、S、F 等掺杂过程,而图4(b)显示了掺杂可能引起的石墨化结构变化。可进行非金属杂原子掺杂的元素主要集中在图4(c)红线区域内,由于杂原子的大小和电负性与碳原子的大小和电负性不同,因此能够有效改变材料电荷分布和电子性质,并与掺杂引起的缺陷一同改变材料的化学活性,并提供大量的催化活性中心[86]。
2.1.1 N 掺杂 氮原子是炭材料最常见的掺杂形式。首先,它的大小与碳原子相似,而且电负性为3.04,大于碳原子的2.55。氮掺杂到炭(NC)中意味着由于氮的电负性较高,更多的电子被吸引到NC中,从而提高了电导率。此外,氮掺杂还在NC 上产生额外的活性中心。当氮与sp2碳骨架中的碳原子键合时,氮的存在形式主要包括以下四种[87]:吡啶、吡咯、石墨化和吡啶-氮-氧化物。其中,吡啶氮是sp2杂化的,它取代了六方环中一个缺陷的碳原子,并为π 体系贡献了一个p 电子。吡咯氮指的是一种sp3杂化的氮原子,它取代了五元环中有缺陷的碳原子,并对π 体系提供了两个p 电子。sp2石墨化氮取代了六方环中的碳原子。吡啶-氮-氧化物表示氧化吡啶-氮键附加氧原子。对于以上氮掺杂结构,确定催化活性位点至关重要,采用Ar+蚀刻技术在掩模上刻蚀出边缘图案的表面,用明确的共轭π 键和严格控制的N物种掺杂来修饰高度定向的热解石墨(HOPG),在所有N 构型中,带有一对孤电子的吡啶氮能与CO2结合,将第一次质子-电子耦合转移形成的关键中间体*COOH,进一步发生质子电子耦合,经*CO 还原为CO,相比之下,石墨氮的电子位于π反键轨道,不容易结合CO2。此外,弱酸性CO2分子优先吸附在吡啶氮位点,也促进了CO2还原优先发生在吡啶氮而不是石墨氮位点[88]。
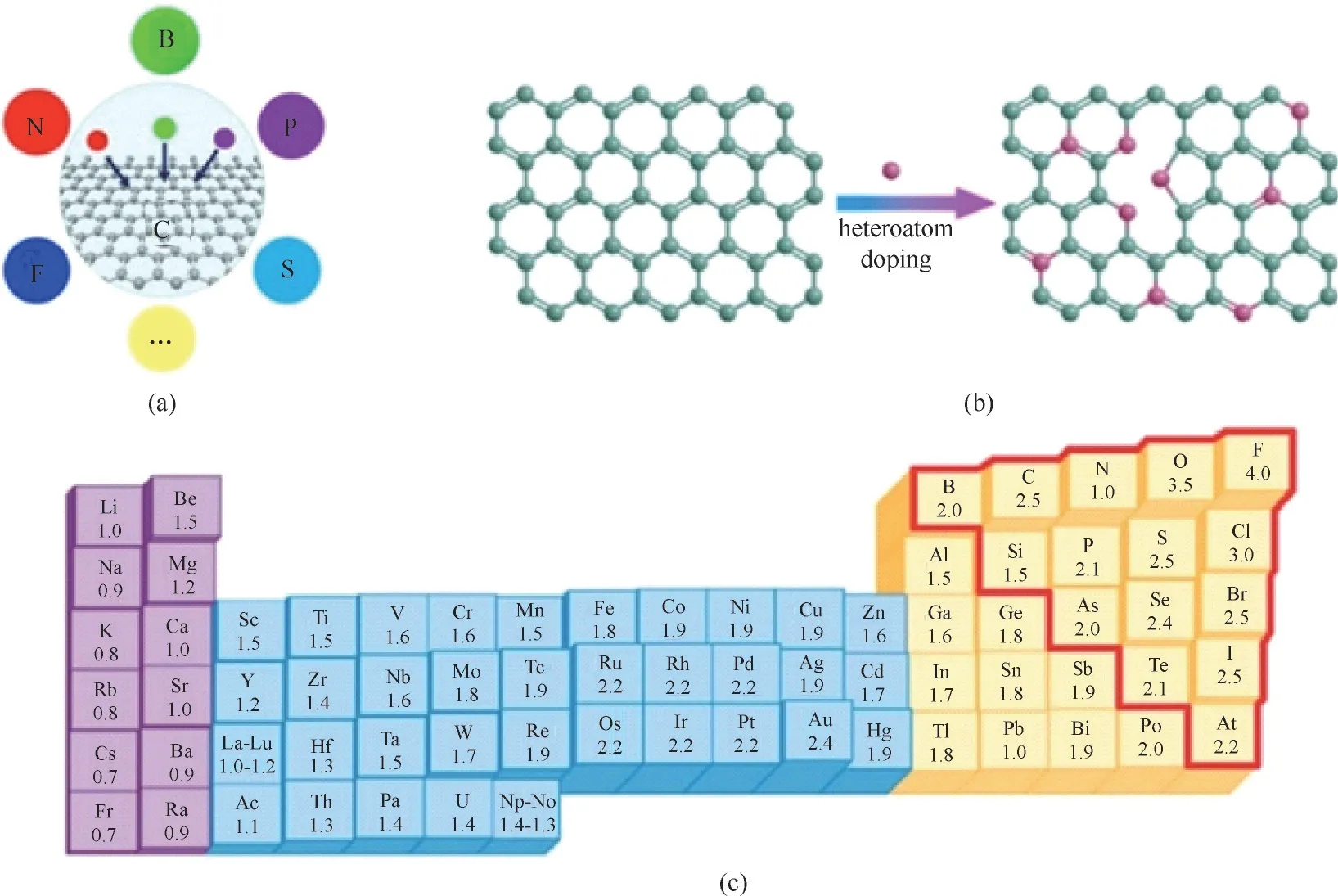
图4 杂原子掺杂炭结构(a)及掺杂方法(b),杂原子电负性(c)Fig.4 Doping(a)and doping method(b)of graphitic carbon structure with heteroatoms,and the corresponding electronegativity of elements(c)
氮掺杂炭基催化剂主要包括掺氮碳纳米管、氮掺杂纳米金刚石等。Kumar 等[89]采用二次掺杂工艺,制备了具有丰富吡啶氮的介孔炭材料,提高了CO2还原的活性与选择性。在-0.58 V(vs RHE)的电位下,法拉第效率为92%、CO 电流密度为6.8 mA·cm-2。Meyer 等[90]通过对涂覆多壁碳纳米管的玻碳电极(CNT/GC)进行氨等离子体处理,得到掺氮碳纳米管。随后通过浸涂聚乙烯亚胺(PEI)、水洗,得到PEI 膜包裹的氮掺杂的碳纳米管。PEI 对CO2具有很高的吸附能力和选择性,起到共催化作用,降低了CO2还原制备甲酸的过电位(400 mV),提高了法拉第效率(87%)和电流密度(9.5 mA·cm-2)。在PEINCNT 中的PEI 层首先可以在NCNT 催化剂周围吸附聚集CO2,并进一步将CO2还原为稳定中间体CO2·—,但是,缺乏对活性中心的深入分析。Wu 等[91]采用化学气相沉积法,在850℃下,Ar/H2气氛中,利用乙腈和二氰胺前体,合成了掺氮碳纳米管,富含吡啶氮(1.1%,原子分数)和石墨化氮(3.5%,原子分数)。电催化性能显示,在0.18 V 过电位下达到了80%的选择性。Liu 等[92]提出了将氮掺杂纳米金刚石/硅棒阵列(NDD/SiRA)应用到电催化CO2还原领域。NDD/SiRA 可以优先和快速地将CO2转化为乙酸,起始电位为-0.36 V(vs RHE),突破了C2产品选择性低的不足,在-0.8~-1.0 V(vs RHE)时,法拉第效率为91.2%~91.8%。
随后,Wu等[93]将具有丰富本征缺陷的碳纳米结构与后处理氮掺杂技术结合获得石墨烯负载型氮掺杂石墨烯量子点(NGQD 结构)[图5(a)]。如图5(b)所示,由此产生的NGQDs 的吡啶氮浓度为3.9%(原子分数),与原始石墨烯相比,NGQDs 具有较小的横向尺寸,暴露更多的边缘位置,获得了高含量的吡啶氮活性中心。在-0.74 V(vs RHE)电位下,CO2还原法拉第效率达到90%,产物主要为碳氢化合物和含氧化合物[图5(c)]。特别是,C2产物乙烯(C2H4)和乙醇(C2H5OH),在-0.86 V(vs RHE)下,C2H4的电流密度为46 mA·cm-2,C2H5OH 的电流密度为21 mA·cm-2,超过了广泛研究的铜催化剂[图5(d)]。
Jhong 等[94]将多壁碳纳米管负载的类石墨型氮化碳(g-C3N4),在1000℃下热解形成氮掺杂炭包覆的碳纳米管纳米复合材料(CN/MW CNT),CN/MW CNTs 催化剂CO 的选择性高达98%,在流动电解池中使用1 mol·L-1KCl作电解液时,CO 部分电流密度可达90 mA·cm-2,比最先进的Ag 纳米粒子高三倍。此外,催化稳定性好,在50 h 内选择性与电流密度无衰减。Song 等[95]报道了一种氮掺杂有序介孔炭,氮杂原子和圆柱形介孔的协同效应促进了质子-电子转移,加速了关键CO-中间体的C—C 偶联,显著促进CO2电还原生成乙醇产物。作者认为有序介孔炭的高比表面积、均匀介孔孔道、突出的化学稳定性和高电导率等优点共同增强了CO2电还原性能。在-0.56 V(vs RHE)的低电位下,CO2转化为乙醇的法拉第效率高达77%。
2.1.2 S 掺杂 Duan 等[88]根据DFT 计算,认为S 掺杂石墨烯中有六种类型[96]:吸附在石墨烯表面的硫(S1),取代边缘碳的硫(S2 和S3),在边缘形成硫/硫氧化物(S4 和S5),以及连接两个石墨烯片的含硫环(S6)。作者认为在石墨烯表面吸附硫(S1)具有最稳定的结构,S2~S4 类型位于锯齿形边缘时能量最低,这种锯齿形缺陷有利于硫掺杂。尽管硫(2.58)与碳(2.55)具有相似的电负性,但是硫原子与碳原子相比,硫的原子半径更大,极化率更高。因此,硫掺杂的炭材料具有较高的自旋密度、边缘应变和电荷离域化等优点。
例如,Pan等[97]提出了一种化学双掺杂方法来调整氮-碳的局部电子环境,认为共掺杂催化剂的活性增强来自于氮和次原子之间的协同效应,探讨了利用硫掺入促进CO2在氮掺杂炭上还原为CO 的方法。作者通过简单的二维结构氮化碳模板热解策略合成了具有N/S 掺杂的二维结构,即N-C 材料和NS-C 材料[图6(a)、(b)]。在490 mV 的低过电位下,N、S 共掺杂的NS-C 催化剂的CO 的法拉第效率为92%、电流密度为2.63 mA·cm-2。作者通过对比单纯硫掺杂的炭材料,发现其只表现析氢活性,从而确认了N、S共掺杂对于CO2还原的协同催化性质[图6(c)]。
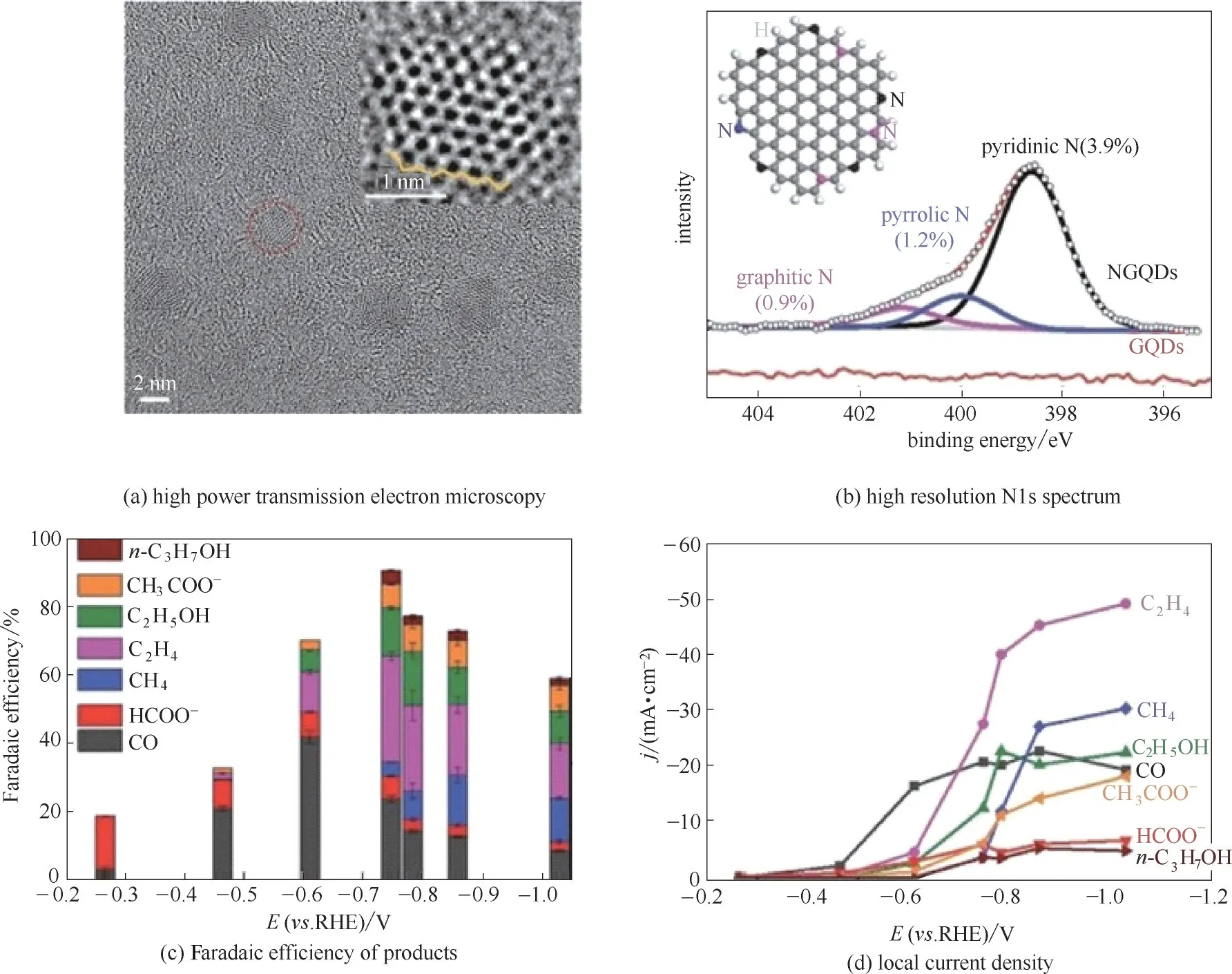
图5 石墨烯负载型氮掺杂石墨烯量子点形貌以及CO2还原电化学测试Fig.5 Morphology of graphene-loaded nitrogen-doped graphene quantum dots and electrochemical measurement of CO2 reduction
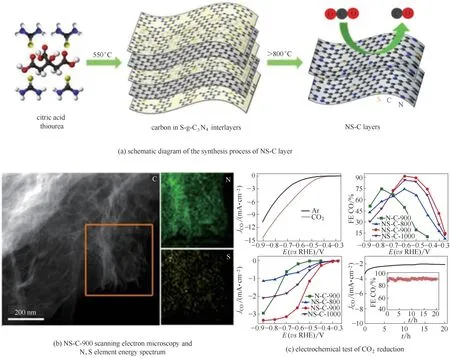
图6 NS-C层的合成过程、形貌以及CO2还原电化学测试Fig.6 Synthesis processes of NS-C layers,surface morphology and electrocatalytic test towards CO2 reduction
2.1.3 B 掺杂 鉴于CO2在氮化硼管上易于被吸附,同比类推到了掺硼金刚石电极上进行CO2电还原的研究[98]。碳骨架中的硼掺杂原子被sp2杂化用于面内掺杂,由于硼原子和碳原子之间的成键能力,保留了石墨烯的平面结构[83,88]。硼掺杂石墨烯有两种面内构型:石墨化硼和硼硅烷。前者是指石墨烯晶格中的碳原子被硼原子取代,后者是指位于π共轭体系中的硼原子。由于硼原子和碳原子之间的电负性差别较大,硼掺杂在碳骨架中引起电荷极化往往使CO2的氧原子稳定下来,从而增强CO2在炭表面的化学吸附。Sreekanth等[99]首次报道了硼掺杂石墨烯(BG)对CO2还原成甲酸的电催化活性。他们通过在Ar 气氛中在900℃下热处理氧化石墨烯(GO)和硼酸的[1∶5%(质量)]混合物制备了硼掺杂石墨烯。作者认为硼的存在引入了不对称电荷,改变了自旋密度分布,从而导致了较高的自旋密度,B 原子和C原子的正自旋密度表明这两个原子都具有催化活性。作者通过DFT 计算,发现硼掺杂打破了离域电荷分布,进而形成不对称的自旋密度分布。B 原子和C 原子都表现正自旋密度,有利于CO2经质子电子耦合生成的*COOH中间体的化学吸附。
Nakata等[100]介绍了掺硼金刚石(BDD)电极,利用海水作为电解质,电化学还原CO2生成甲醛。与sp2碳结构相比,sp3碳结构表现出多电子还原活性,硼掺杂金刚石(BDD)可以通过4e-转移途径将CO2还原成甲醛(HCHO),sp3键合硼(B-sp3)在HCHO 生产中起着关键作用,随着BDD 电极中B-sp3含量的降低,HCHO的法拉第效率急剧下降。该方法显著抑制了析氢活性,促进了高阶产物的生成。BDD 电极对甲醛表现出非常高的法拉第效率(74%),稳定性很好,恒电位电解20 h 保持稳定。张宇晶[101]采用热丝化学气相沉积镀膜设备,以超低阻硅为基底,以CH4为碳源、B2H6为硼源、N2为氮源,通过调控反应参数成功制备了一系列不同硼氮掺杂浓度的BND 膜电极。通过进行电催化还原CO2测试得到优异的电催化活性,乙醇的法拉第效率可达93.2%。线性伏安曲线表明,BND 膜电极具有较低的析氢电位(1.62 V vs Ag/AgCl),可降低析氢反应对CO2还原的竞争,提高电流效率;提高BND 中氮的掺杂量,电极的阴极电流起始电位由-1.71 V 变为-1.27 V,表明氮的掺杂有利于降低反应的能耗。Liu 等[102]采用化学气相沉积法在硅衬底上沉积BND 膜,发现存在类似的规律。由于硼和氮共掺杂的协同效应,BND 电极上显示了良好的乙醇选择性,其法拉第效率达到93.2%(-1.0 V vs RHE)。
2.1.4 P 掺杂 磷掺杂多孔炭是另一种具有代表性的电催化CO2还原活性的材料。DFT 计算表明,关键中间体COOH*在P—C 键上的结合能高于P—O键。此外,P—C 键还能改善电子转移,是CO2还原的活性位点。目前的工作集中于解析P—C 构型的影响及磷掺杂炭催化剂的结构与性能优化。例如,Liu 等[103]分别采用浸渍法(IP)和化学气相沉积法(CVD)制备了掺磷洋葱状炭(OLC),分别记为POLC-IP 和P-OLC-CVD,其中P-OLC-CVD 包含等量的P—C 和P—O 键合,而P-OLC-IP 只包含P—O键合。作者发现,COOH*的化学吸附在P—C 和P—O 键与磷相邻的碳原子,根据DFT 计算,COOH*对P—C 键的吸附强于对P—O 键的吸附。相比于P—O 键,P—C 键合可以提高COOH*中间体的稳定性,提高了催化性能。P-OLC-CVD 和P-OLC-IP 的CO法拉第效率分别为81%和60%。
尽管无金属杂原子掺杂炭基催化剂可以突破传统金属催化材料的线性关系的限制,但是目前开发的非金属多孔炭催化剂仍有很大改进空间。大部分非金属掺杂的炭材料是通过2e-转移途径催化CO2还原为CO 或HCOO-。虽然,少量炭基催化材料可得到C 数≥2 产品,但其法拉第效率还是低于金属催化剂。此外,材料合成相对复杂,成本也相对较高。例如,掺杂的金刚石对C2H5OH 具有很好的选择性,但等离子体或CVD 法合成这类材料制备过程复杂。此外,杂原子掺杂炭模型催化剂的C—C 偶联机理仍然有待阐明。与金属催化剂相比,在相同的测试条件下,炭催化剂的CO2还原的电流密度通常比较低。
2.2 过渡金属-氮-炭(M-N-C)电催化材料
针对非金属杂原子掺杂炭材料在CO2电催化还原中的局限性,研究人员发现进一步引入过渡金属,可显著提高CO2电催化的电流密度[104]。不同的载体性质导致过渡金属-载体相互作用差异[105-106],因此过渡金属修饰氮掺杂多孔炭的形式也各不相同。研究发现,M-N-C 类型催化剂的活性中心为与氮配位的过渡金属(M-Nx)[107];也有研究认为与石墨烯缺陷位点锚定的Ni原子是活性中心,而氮的存在促进了缺陷位点的创制[108]。下面按过渡金属物种(纳米颗粒及单原子)、过渡金属单原子的顺序进行综述。
2.2.1 过渡金属修饰的氮掺杂多孔炭 过渡金属-氮共掺杂的多孔炭材料(M-N-C)最初被发现是优异的氧还原催化剂。近年来,该类型催化剂在CO2电催化还原中表现出高活性与选择性,尤其是电流密度显著提高[109-110]。起初研究人员借鉴氧还原催化剂的制备方法,过渡金属(Ni、Fe 等)的负载量通常高于10%(质量分数),在热处理过程中,这些过渡金属物种促进了催化石墨化,在金属颗粒表面生长了一层致密的石墨化炭。因此在酸洗过程中,这些包覆的过渡金属颗粒很难除去,在一定程度上贡献了析氢活性[111]。例如,Varela 等[79]在氮掺杂多孔炭中引入过渡金属Fe 和Mn,发现CO2还原制备CO 活性显著提高。在所研究的电位范围中,M-N-C 材料都表现出CO2到CO 的还原活性和析氢活性;有趣的是,FeMn-N-C 和Fe-N-C 材料还检测到微量的CH4生成。作者认为这是由于不同活性中心的CO 吸附能差异所致。
随后,Ju 等[112]基于一系列M-N-C(M=Mn,Fe,Co,Ni,Cu)系统研究了CO2还原的活性中心及催化机制[图7(a)],发现Fe-N-C 和Ni-N-C 结构,特别是Ni-N-C 结构,其CO 选择性可以与Au 基和Ag 基催化剂相当[图7(b)]。作者以配位M-Nx活性中心为模型,通过DFT 理论,确定了三个不同反应动力学的电势区[图7(c)]。区域1:低过电位区,CO 产生的起始电位与反应中间体COOH*的结合能有关,该区域模拟结果与电化学测量一致,催化剂分为两组:Co,Fe 和Mn 只需要较小过电位,而Ni-Nx和Cu-Nx结构是与COOH*结合最弱的单中心,因此需要最大的起始过电位。区域2:中等过电位区,-0.6 V (vs RHE)处的CO周转频率与CO*的自由能呈线性相关,这表明速率控制中间体已从COOH*转向CO*。区域3:高过电位区,每种M-N-C 催化剂在-0.8 V (vs RHE)处析氢反应(虚线路径)和CO2电催化反应(实线路径)的自由能图,从该图可以看出Fe、Co和Mn基催化剂开始强烈催化析氢反应(H++ e-→H*→H2(g))。作者从原子尺度对M-Nx活性位对CO2还原历程进行了解析,为设计M-N-C 类型CO2还原催化剂提供了指导。此后,涌现了大量的M-N-C 类型催化剂,对于催化机理的研究也更为深入。比如Zhang 等[113]的一项研究表明,Fe-N4是产生CO 的活性中心,而不是Fe 纳米颗粒或氧化铁。通过进行X 射线吸收光谱(XAS)测试,进一步证明了二价Fe 为活性位点,而非三价Fe[114]。Pan 等[115]也发现了类似的规律,低价金属在低过电位下,对CO2电还原表现出显著的活性。
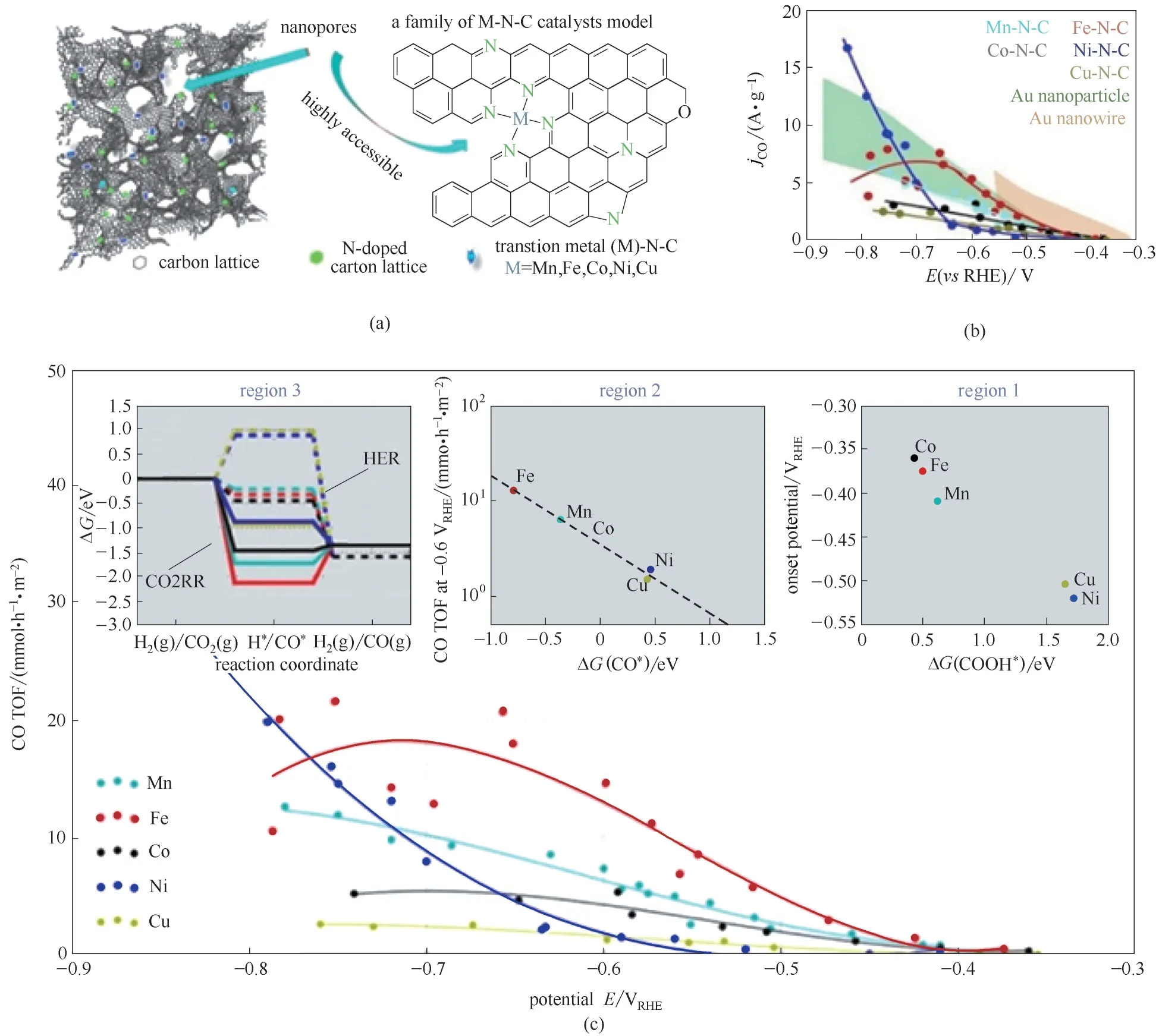
图7 M-Nx材料模型与局部结构示意图(a),与Au基催化剂相比,M-N-C催化剂的质量归一化CO部分电流(质量活性)(b)和实验与模拟的相关性(c)Fig.7 Model and a schematic local structure(a),and catalyst mass-normalized CO partial currents(mass activity)vs applied potential compared to state-of-art Au catalysts(b)and experimental correlation to simulations(c)for M-N-C catalysts
2.2.2 过渡金属单原子修饰的氮掺杂多孔炭 随着材料制备技术及表征技术的不断提高,单原子催化剂(SACs)在材料科学领域受到了关注,并为电催化CO2还原提供了模型催化剂[116-117]。单原子催化剂由于具有催化活性高、稳定性好、选择性好、原子利用率高等优点,在CO2电催化中具有重要的应用前景[118]。例如,Yang 等[119]报道了一种原子分散镍氮化石墨烯催化剂(图8),他们所制备的催化剂具有二维结构,表面光滑[图8(a)~(c)],并比较了不含硫前体和添加硫磺前体的单镍原子催化剂[图8(d),分别表示为A-Ni-NG 和A-Ni-NSG]。根据XAS 和XPS[图8(e)、(f)],确定具有d9电子构型的一价Ni(Ⅰ)原子中心为催化活性中心。结果证明Ni 3dx2-y2轨道中未配对电子的非局域化和从Ni(Ⅰ)到CO2中的2p 轨道的自发电荷转移,形成了CO2δ-物种,这些过程降低了活化CO2所需的能量。单Ni原子催化剂具有较高的CO2还原活性,当过电位为0.61 V(vs RHE)时,催化剂质量比电流为350 A·(g catalyst)-1,周转频率为14800 h-1,CO 法拉第效率为97%。在电流密度为22 mA·cm-2的条件下,连续反应100 h 后,催化剂的初始活性保持了98%。
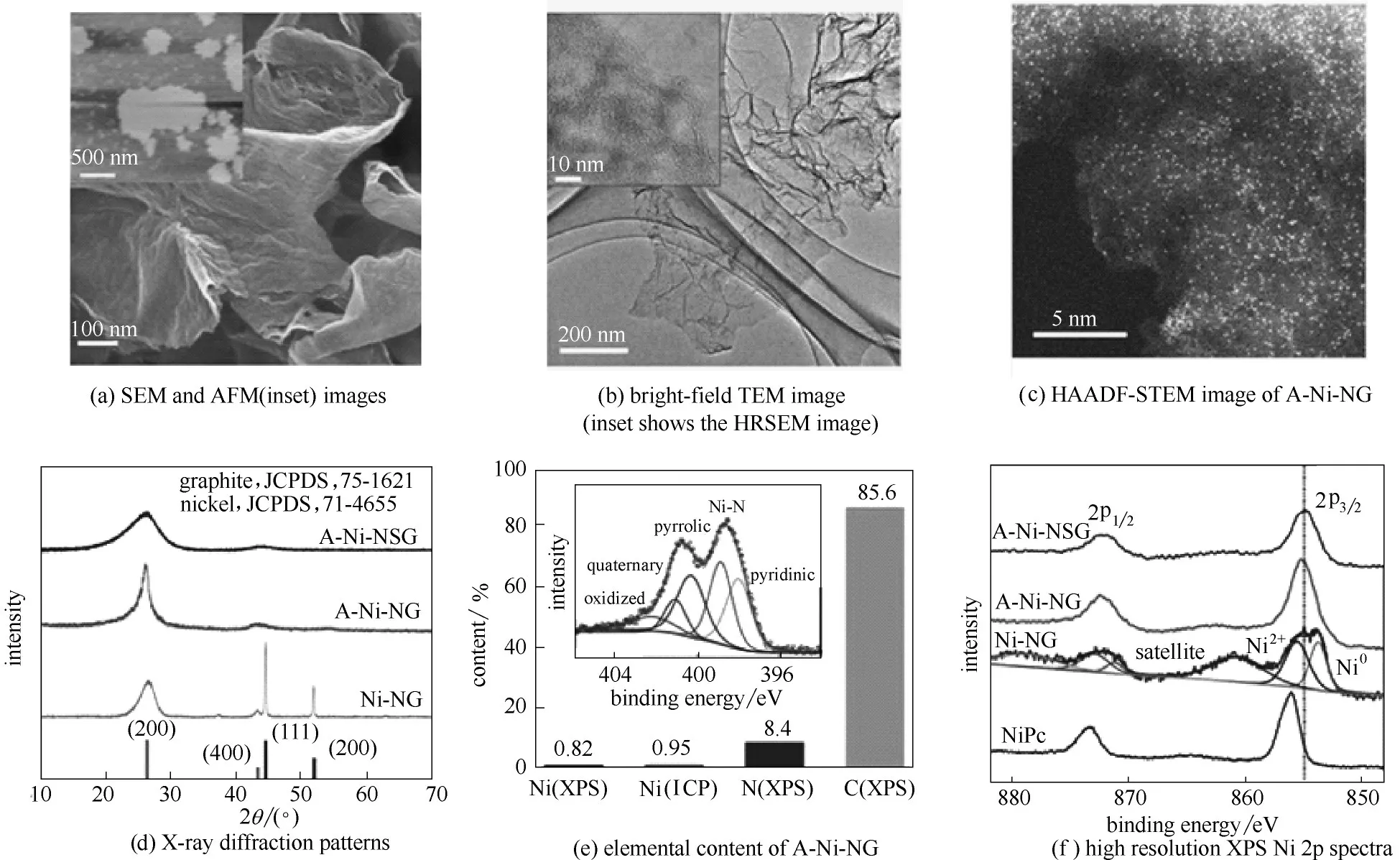
图8 分散在氮化石墨烯上的单个镍原子的结构表征Fig.8 Structural characterization of single nickel atoms dispersed on nitrogenated graphene
此外,单原子Cu-N-C 催化剂可以将CO2转化为高值化学品,如碳氢化合物或醇类。比如He等[120]提出了一种大规模合成单原子Cu 负载氮掺杂纳米炭纤维催化剂(CuSAs/TCNFs)的策略。结果表明,该CuSAs/TCNFS 膜具有优良的力学性能,可直接用作CO2还原的阴极,在液相中可产生近44%法拉第效率的甲醇。根据DFT 计算,Cu-Nx位点表现出较高的结合能,促进了*CO中间体进一步加氢得到甲醇。同时,要生成CH3OH,应将*COH 中间体还原为*CHOH,而不是*C,因为是生成CH4的关键步骤。根据DFT 计算(图9),Cu-N4结构上具有中等自由能(约0.86 eV)。相反,*的自由能(约1.88 eV)比CH3OH 生成的任何一步都要高。因此,CuSAs/TCNFs 催化剂上的单原子Cu 位点倾向于生成CH3OH 而不是CH4。因此,研究不同配位环境下不同过渡金属原子的电子性质,探索将单个原子结合到合适的基体材料中的有效方法,可以为利用这些低成本材料开发高选择性和高值化学品CO2电催化剂提供新的见解和设计原则[74]。
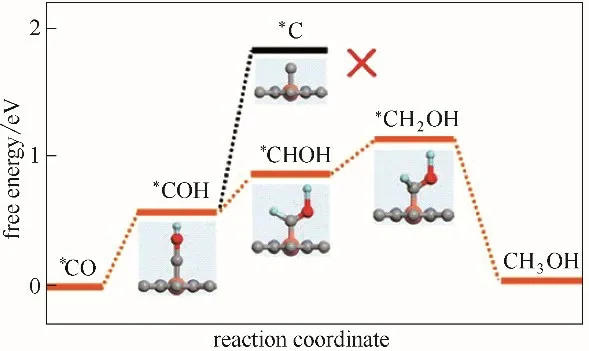
图9 Cu-N4结构上*CO转化为CH3OH的自由能(橙色、灰色、红色和浅蓝色球体分别代表Cu、C、O和H原子)Fig.9 Free energies for conversion of*CO to CH3OH on Cu-N4 structure(orange,gray,red and light blue spheres stand for Cu,C,O,and H atoms,respectively)
2.3 过渡金属配合物负载型多孔炭催化材料
分子催化剂具有明确的活性中心和精确的可调控结构,作为均相催化剂在CO2电催化还原中也得到了广泛的探索。如何融合均相催化剂及多相催化剂的优势成为CO2电催化领域研究热点。一方面可对活性中心精确设计,另一方面可利用炭载体的导电性,同时提高催化剂的可操作性。例如,Wang 等[121]在多壁碳纳米管表面用Co(Ⅱ)-四氢吡啶配合物改性,在中性水溶液可将CO2转化为CO,在过电位为-0.35~-0.58 V(vs RHE)时,法拉第效率可达100%,过电位低,过电位在240~440 mV间,电流密度可以维持1~19.9 mA·cm-2[122]。
近期,Wu 等[123]进一步创新了负载型配合物催化剂的设计,将酞菁铁(FePc)、酞菁钴(CoPc)和酞菁镍(NiPc)大分子负载在碳纳米管(CNTs)上,合成了配合物/炭催化剂。在0.1 mol·L-1KHCO3水电解质中,三种MPc/CNT(M=Fe,Co 或Ni)催化剂在中等过电位范围内均表现出良好的CO 生成选择性, 当过电位较 高 时,NiPc/CNT 和FePc/CNT 的H2选 择 性 高 于CO,而CoPc/CNT 生成甲醇[MeOH,图10(a)],并且甲醇是唯一液相产物。CoPc/CNT 催化生成甲醇的起始电位为-0.82 V(vs RHE)。在-0.94 V 处,达到最大法拉第效率44%,最大部分电流密度为10 mA·cm-2。该类型催化剂实现了CoPc 活性组分在碳纳米管上的分子级分散[图10(b)、(c)]。然而,CoPc/CNT 的电解稳定性还有待进一步改善,CO2电催化制MeOH在5 h 后甲醇的法拉第效率下降到0.6%。作者推测催化剂失活是由酞菁配体还原引起的。针对此,作者进一步做了改善,在酞菁环上添加给电子的氨基取代基。结果发现,改进后的催化剂在12 h 内未发现明显衰减。氨基取代提高了催化剂的耐久性,证明了配体工程可在提高杂化分子体系催化性能方面发挥作用。这与Li 等[124]利用一系列分子催化剂实现CO2到乙烯的发现吻合。
2.4 贵金属修饰氮掺杂多孔炭催化材料
虽然贵金属存在储量有限、价格高等问题,但是通常表现出较好的催化活性。随着单原子催化剂的发展,在极大程度上降低贵金属的用量,也受到了研究者的广泛关注。例如,Wang等[125]研究了在成本效益较高的过渡金属碳化物(TMC)上负载钯,以减少Pd 的使用并调节CO2电催化还原生产合成气的活性和选择性。碳化钽(Pd/TaC)表现出比Pd/C更高的CO2电催化活性、稳定性和法拉第效率,同时大大降低了Pd 的负载量。原位测量证实在CO2还原环境下Pd 转变为氢化物(PdH)。DFT 计算表明,过渡金属碳化物底物会改变反应关键中间体的结合能。这项工作表明使用过渡金属碳化物底物作为低成本和稳定的底物来修饰Pd 以增强CO2电催化还原活性具有光明的前景。Liu 等[126]通过创制富含表面缺陷的炭材料载体,制备了Pt 单原子分散的高效电催化剂,在氧还原中表现出与商业Pt/C 相当的表观电催化性能。其中,双缺陷中四个碳原子锚定一个Pt 原子所构成的Pt-C4被确认为活性中心,上述Pt 单原子分散多孔炭催化剂结构设计与活性位创制策略也适用于电催化CO2还原。
3 结论与展望
电催化CO2还原制备高附加值的化学品是目前的研究热点,非贵金属多孔炭基CO2电催化材料取得一系列突破性进展。然而为了实现工业水平应用,还需要在以下几个方面做更多的积累。
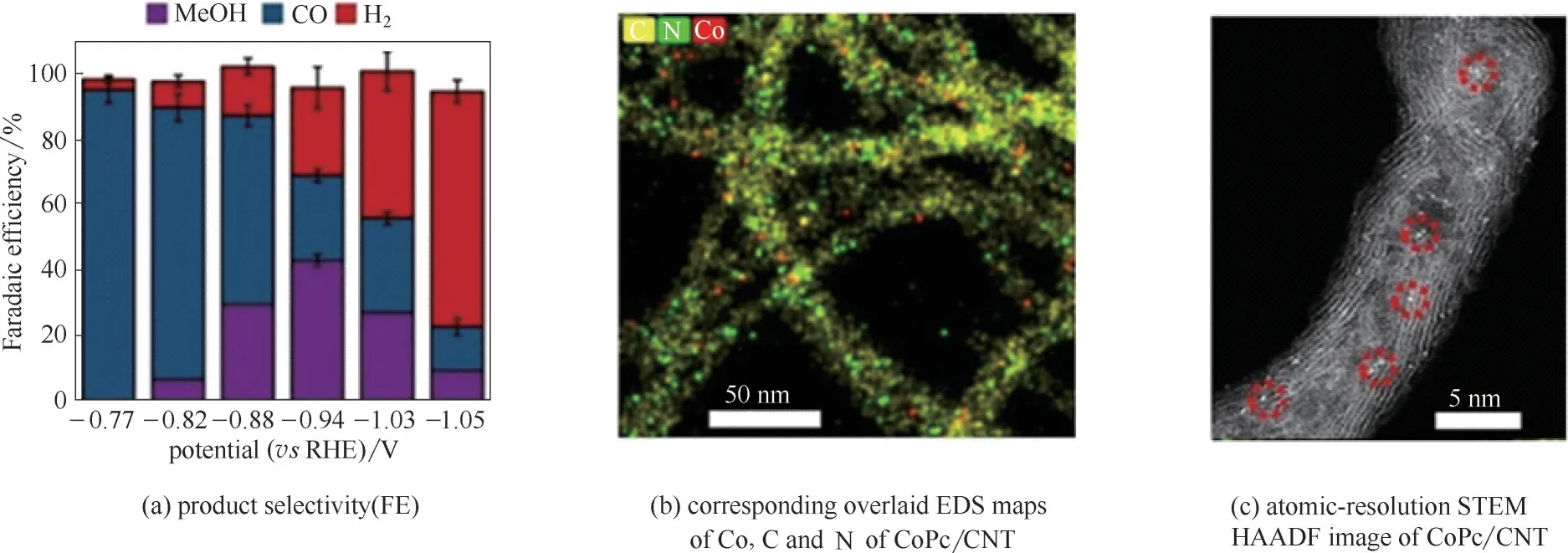
图10 碳纳米管负载的CoPc分子对CO2还原的催化性能和结构表征Fig.10 Structural characterization and catalytic performance of CoPc molecules supported on CNTs for CO2 reduction
(1)提高催化剂的活性及选择性。对于电催化CO2制CO,目前进展最好,选择性可达到90%以上。虽然非金属纳米金刚石修饰的氮掺杂石墨烯催化剂可以得到乙烯或乙醇,过渡金属-氮-炭(比如Cu-N4修饰的炭纤维)催化剂可以得到甲醇,但是,可以得到高阶碳氢化合物或者含氧化合物的催化剂仍然较少。因此,发展新结构炭基催化剂实现高选择性制备高阶产物仍然是未来研究的重点之一。此外,高效筛选合适的电解质也是需要关注的方向。目前应用最广泛的电解质是KHCO3与KOH,对于二者的研究也较多。在KHCO3电解液中,CO2的溶解性较低,达不到工业水平电流密度(200 mA·cm-2)。对于KOH 电解液,KOH 易与CO2反应,因此不能直接与CO2接触。在电解质筛选上,离子液体由于具有良好的导电性、低挥发性、较宽的电化学稳定电位窗口等优点,在CO2电催化领域将发挥更大的协同作用。
(2)定向制备高密度的活性位。对于多孔炭基催化材料的活性位认识已有大量报道,但由于多孔炭材料结构的复杂性,尚缺乏精准解析手段。广泛认为非金属杂原子掺杂位点是CO2还原的活性位,但是很难排除痕量的金属杂质对催化活性与选择性的贡献。过渡金属氮配位中心(M-Nx)被认为是高效催化活性中心。然而,活性中心的浓度还相对较低。因此,发展具有高浓度单一活性位点的模型催化剂制备技术,并结合原位谱学表征是未来研究的重点。值得提出的是,过渡金属单原子-氮-炭是一个较为理想的催化剂模型;此外,利用过渡金属配合物结构精准调控的优势,发展炭负载型催化剂也是有前景的方向。
(3) 技术与经济性分析。①目前工业上CO2催化转化仍然是热催化占主导。CO2电催化实际应用的最大障碍是能量效率低,经济上不合算,特别是产物为高值化学品时,选择性、过电势、电解能量效率低多需要改善。通过系统的技术与经济性分析,发现在目前条件下(催化剂、电解质)具有高选择性的两电子转移产物(如CO2到CO,进一步经合成气通过费托合成制备高值化学品)是最有可能盈利的方向。②由于反应物扩散限制,接触到的有效活性位密度低,催化电流密度远没有达到工业水平。通过创新催化剂结构及电极构型(如气体扩散电极),提高催化效率的研究仍然有待加强。③另一个解决思路是,发展阳极电合成,与阴极CO2还原构成配对电解池,最大程度提高能量利用率,发展可以实用的电解体系。
