DFT模拟铂催化肼的分解过程
2020-06-21唐菲菲张晓腾石伟群夏良树
唐菲菲,郝 帅,张晓腾,石伟群,夏良树,*
(1.南华大学 核科学技术学院,湖南 衡阳 421001;2.中国科学院 高能物理研究所 核能化学实验室,北京 100049)
随着人类社会的发展,能源短缺问题日益突出,核能具有转换效率高、稳定可控、对环境友好等优点,有着广阔的市场空间和发展前景。其中,在乏燃料的处理问题上,目前有闭式循环和一次通过式两种方式,我国采用的是闭式循环。在闭式循环的后处理工业流程中,还原剂的选择对铀钚分离十分重要,U(IV)—肼为还原剂的1B工艺较为成熟稳定,是目前最为理想的还原剂。然而U(IV)不稳定,在硝酸环境下很容易被氧化为U(VI),故实际应用中U(IV)需要有单独的制备工艺。
目前制备U(IV)的工业化流程有电解法和催化还原法,电解法有过程简单、电流效率高等特点,在后处理厂已有生产规模的应用。但放射环境中工业规模的隔膜使用、处理,电极的更换和再生等都存在困难,因此催化还原正在逐渐的取代直接电解法,其中以铂催化肼还原U(VI)制备U(IV)的研究较多。国外Swanson[1]、Boltoeva[2-4]、Abdunnabi[5]和国内李斌[6,7]等人研究了不同体系下温度、酸浓度、肼浓度、催化剂用量等因素的影响。
虽然大多学者都认同酸性条件下肼的催化分解是异相催化分解,但对肼异相催化分解的具体机理并没有形成一致观点。肼作为一种表现良好的航天燃料,目前对于其分解的研究大多在航天领域,主要研究其分解制氢,肼的各种催化剂,肼及衍生物的分解等[8-15]。在涉及铀、钚等放射性元素的后处理方向,对肼的研究则很少,且研究以实验居多[6,7,16-19]。
由于实际反应过程较迅速,且实际反应环境为酸性溶液环境,对过渡态的捕捉、结构研究受到一定限制,故从实验角度探究肼催化分解的具体机理存在困难。基于目前实验数据的不足,本文从量子化学角度出发,以模拟计算的方式对Pt催化N2H4分解反应进行密度泛函研究(DFT)。DFT是较为新颖的一种理论方法,近年在几何构型、反应机理探究等方面有广泛的应用[20-26]。通过模拟计算中性、酸性溶液条件下N—N以及N—H两种断键过渡态结构,对各结构进行几何优化,探究Pt催化N2H4分解反应机理。
1 模拟计算
1.1 计算方法
采用密度泛函理论中的杂化泛函B3LYP,对Pt原子采用Los Alamos赝势基组Lanl2TZ(f),体系中的其他原子则采用6—31 G**基组,Pt原子为原子态,采用smd溶剂化模型,在中性溶液条件下进行模拟计算。实际的催化分解过程中常添加酸溶液,因此通过下列两种方式探究H+的影响:质子化N2H4,或添加H3O+。
采用ts方法寻找过渡态,确定结构有且只有一个虚频,且该虚频下的震动模式符合预设断键趋势。对过渡态结构进行IRC验证,确定IRC曲线平滑完整,对两端端点结构进行几何优化以确定过渡态与反应物和产物相连。对得到反应物、过渡态和产物的几何结构、热力学数据等进行分析,以此判断反应能垒,进而探究Pt催化N2H4分解的反应机理。
所有计算采用Gaussian09程序[27]。
1.2 结果与讨论
计算得到过渡态结构如图1所示,TS1为中性溶液条件下N—N断键的过渡态,TS2为N2H4质子化条件下N—N断键的过渡态,TS3为H3O+条件下N—N断键的过渡态;TS4为中性溶液条件下N—H断键的过渡态,TS5为H3O+条件下N—H断键的过渡态,TS6为H3O+条件下N—H断键逆过程的过渡态。

图1 Pt催化N2H4分解所有过渡态结构
1.2.1 铂催化肼N—N断键模拟计算
根据图1所示的N—N过渡态结构(TS1、TS2、TS3),对其IRC曲线两端端点结构分别进行几何优化,分别得到三种过渡态对应的反应物和产物结构(见图2)。根据图中Re1(反应物1,下同)、Re2、Re3、P1(产物1,下同)、P2、P3可见,TS1、TS2、TS3结构连接着N2H4分子N—N断键的反应物及反应产物。由此证明计算得到的结构为N2H4在Pt催化下N—N断键的过渡态结构。根据TS1、TS2、TS3结构可得其几何数据如表1所示。




图2 N—N断键反应物、产物结构

表1 N—N断键过渡态结构几何数据

TS3与TS2相比,H2O对结构造成了较大影响。根据表1数据可知,TS3中∠2H—1N4N—5H和∠3H—1N4N—7Pt相较TS1、TS2有巨大变化,由侧视图3可见TS3结构与TS1、TS2结构存在明显差异。TS1与TS2结构中,4N被1N遮挡。TS3中4N被1N遮挡,5H被8H遮挡,6H则被3H遮挡。由TS1结构可见,以4N—7Pt为轴,2H、5H与3H、6H分别位于N—Pt轴两侧,具有一定对称性。TS2结构中,受质子化8H的影响,2H、3H发生了轻微的偏转,但2H、5H与3H、6H依然位于N—Pt轴的两侧。而TS3中,质子化的8H受到H2O的影响,虽然1N—8H键长未产生明显变化,但O原子的吸引还是带动8H产生了较大偏转,并使得2H、3H也一并发生偏转,使得TS3较TS1、TS2结构发生较大变化。
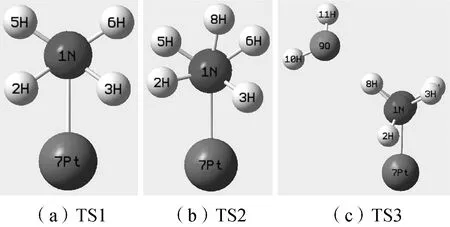
图3 N—N断键过渡态结构对比图
根据TS1、TS2、TS3、Re1、Re2、Re3、P1、P2、P3结构的热力学数据可得,中性溶液中N—N断键能垒值E=42.16 kcal/mol,质子化影响下N—N断键能垒值E=10.11 kcal/mol,H3O+影响下N—N断键能垒值E=12.64 kcal/mol,其势能如图4所示。可见酸性条件下N—N断键能垒有着明显降低,由此判断酸性条件下N—N断键比中性条件更容易发生。
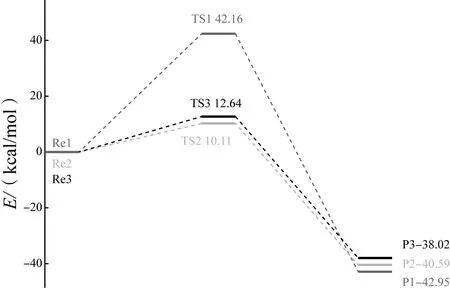
图4 N—N断键势能
1.2.2 铂催化肼N—H断键模拟计算
根据图1所示的N—H过渡态结构(TS4、TS5、TS6),对其IRC曲线两端端点结构分别进行几何优化,分别得到三种过渡态对应的反应物和产物结构,就TS5对应的反应过程而言,还存在中间态IS5(见图5)。根据图中Re4、Re5、Re6、P4、P5、P6可见,TS4、TS5、TS6结构连接着反应物及反应产物。由此证明计算得到的TS4、TS5结构为N2H4分子在Pt催化下N—H断键的过渡态结构,TS6为H3O+影响下N2H4分子N—H断键逆过程的过渡态。






图5 N—H断键反应物、产物结构
根据TS4、IS5、TS5结构可得几何数据如表2所示,由于TS6为N—H断键逆过程的过渡态,故其几何数据单独列出。

表2 N—H断键结构几何数据
由表2可见,1N—2H—3H的·NH2结构较为稳定,基本没有受到H3O+的影响。N—N、4N—7Pt键长在H3O+的影响下也只略有增加。而会发生N—H断键的N—H键长(TS4中为4N—5H,IS5和TS5中为4N—6H)则先发生较大缩减再增大,但TS5结构中的N—H键长依然小于TS4结构。通过对比TS4、TS5的虚频震动模式可知,TS5中5H的震动幅度明显大于TS4中的6H;TS4中的1N和4N基本在N—N连线上进行小幅度的直线震动,而TS5中的4N受H3O+上11H的影响,其震动方向转移为垂直于N—N连线的方向;H3O+的11H则在8O与4N之间进行震动。
由二面角的数据可知,未N—H断键的H原子(TS4中为6H,TS5中为5H)发生了大幅度偏转,如图6所示,图中三个结构的1N均被4N遮挡。以1N—4N—7Pt面为参考,TS4中6H与5H位于参考面两侧,而TS5中5H和6H均位于参考面同侧,且TS4中6H较TS5中的5H更偏离4N—7Pt线。又由图6中IS5与TS5结构可知,在N—H断键过程中,2H、3H较对称分布在4N—7Pt线两侧,随着反应的进行,2H、3H会发生偏转,使得2H更靠近参考面。由TS5键角数据可知,4N—11H—8O三个原子基本处于一条直线上。推断N2H4上的N原子对H3O+上的H+有明显的吸引,使得H3O+逐渐靠近,而H3O+的靠近又影响到N2H4上的H原子,使其向H3O+的反方向发生明显偏移。
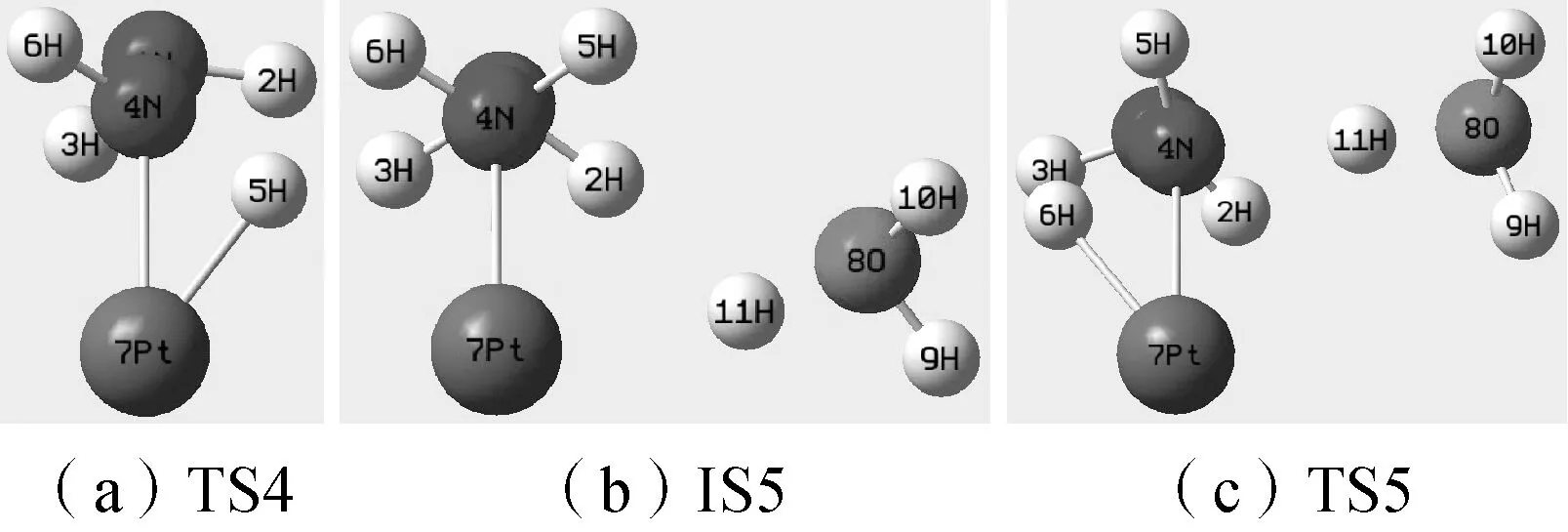
图6 N—H断键结构对比图
TS4、IS5、TS5、Re4、Re5、P4、P5结构的热力学数据可得,中性溶液中N—H断键能垒值E=27.77 kcal/mol,H3O+条件下N—H断键能垒值E=49.82 kacl/mol,可见酸性溶液中能垒有显著升高,势能折线图如图7所示。

图7 N—H断键势能
N—H断键逆反应过程的Re6、TS6、P6几何数据见表3所示。由Re6、TS6与P6结构可知,H2O分子在反应过程中并无太大变化,H3O+对N2H4的影响主要是由Pt吸附的9H造成的。随着5H逐渐被4N吸引,2H、3H以N—N连线为参考线,由本来Pt原子对侧的方向,逐渐偏转至靠近Pt原子的一侧。1N—4N—7Pt的角度也有轻微的增大,H2O分子也逐渐向远离N2H4分子的方向移动。

表3 N—H断键逆过程几何数据
根据Re6、TS6、P6结构的热力学数据可得,H3O+影响下N—H断键逆反应能垒值E=25.47 kcal/mol,与中性溶液中N—H断键能垒27.77 kcal/mol相近。
2 铂催化肼机理
由图2的P1、P2、P3结构可知,酸性条件下N—N断键后的·NH2、·NH3结构,与P1相比有一定的偏转。推测N—H断键的机理如下:中性环境下,N2H4吸附到Pt上,未吸附在Pt上的·NH2结构发生偏转向Pt靠拢,并在Pt的催化作用下发生N—N断键。酸性环境下,由于H2O中9O原子对8H原子的吸引,使得·NH3结构与中性溶液相比发生较大偏转,而8H的影响,使N—N断键更容易发生。
根据TS4、TS5、TS6推测N—H断键的机理如下:中性环境下,N2H4在Pt的催化作用下发生N—H断键,随后Pt吸附·H,此时Pt与·H总体电荷基本为零。由于N—H断键后的·N2H3结构中,N原子带有大量负电荷,Pt原子受其影响带轻微正电荷,继而附着在催化剂Pt上的·H带轻微的负电。N原子和·H带有同电性的电荷,因此·H可以不受·N2H3结构的影响,顺利地从催化剂Pt上解吸下来。酸性条件下,Pt原子吸附H+后,Pt与H+的结构不再呈电中性,而是带了较大的正电荷,此时N2H4上的H原子也带有正电荷,因此N—H断键受到一定抑制。同时N—H断键产生的·H吸附到催化剂Pt上后,也受到带电荷的影响,带上了轻微的正电荷。·N2H3结构中N原子依然带有较大的负电荷,因此会吸引带正电荷的·H,抑制·H从Pt上解吸,同时导致催化剂Pt上未脱落的·H有被·N2H3再次夺回的趋势。也就是酸性溶液环境下,由于H+造成了Pt的电荷正负性的变化,使·H从催化剂Pt上脱落之前,有被·N2H3夺回并最终导致N—H断键后重新恢复的趋势,即发生了N—H断键逆反应,该过程与实验中得到的H+反应级数为负的结论相吻合[7]。
中性条件下N—N断键能垒E=42.16 kcal/mol,N—H断键能垒E=27.77 kcal/mol,由此可见,中性溶液环境中,Pt催化下N2H4更容易产生N—H断键。而酸性条件下,由于质子化的影响,N—N键长被拉伸,使得N—N断键能垒值大幅度降低,E=10.11 kcal/mol。而N—H断键则受到H+抑制作用,不仅N—H断键能垒值升高到49.82 kcal/mol,还可能造成N—H断键的逆反应,使得N—H断键变得困难。
因此,基于N—N断键、N—H断键的能垒值,推断中性溶液中N2H4在Pt催化下N—H断键为主要方式,而酸性溶液中,N—N断键为主要断键方式。
3 结论
(1)通过模拟计算得到了中性、酸性溶液环境下N2H4在Pt催化下N—N、N—H两种断键方式的过渡态结构。
(2)酸性溶液中的N—N断键比中性溶液更容易发生,溶液中H2O的存在会对N—N过渡态构型造成较大影响。
(3)酸性溶液中,H+不仅影响N2H4中N—H断键,使其发生更为困难,还可能造成N—H断键逆反应,对·H的形成有很大影响。
(4)在Pt催化的中性溶液环境下,N2H4更易发生N—H断键,而酸性条件下,则更易发生N—N断键。
