转移性癌性骨痛的分子机制及临床治疗研究进展
2020-06-19综述审校
综述 审校
中、晚期癌症患者均伴有不同程度的疼痛,其中约44%为难以忍受的持续疼痛[1]。癌痛严重影响患者的生存质量并造成沉重的心理负担。在所有癌性疼痛中,肺癌、乳腺癌、前列腺癌、肾癌及甲状腺癌等骨转移引起的骨破坏性骨痛是最为常见的症状[2-3]。转移性癌性骨痛发病机制复杂,包括炎性疼痛及神经病理性疼痛等多种类型,涉及肿瘤细胞、免疫细胞、骨细胞和支配骨骼及骨髓的感觉神经元之间的相互作用。在治疗上除了世界卫生组织(WHO)推荐的三阶梯镇痛方案外,双磷酸盐及神经调控等也被应用于癌性骨痛治疗中。阐明癌性骨痛的分子机制对优化治疗方案具有指导意义,本文将对癌性骨痛的发病机制及临床治疗进展进行综述。
1 癌性骨痛分子机制
1.1 癌细胞和肿瘤基质细胞释放致痛因子
在肿瘤微环境中,癌细胞及多种基质细胞,如内皮细胞、成纤维细胞及免疫细胞,可分泌多种细胞因子及趋化因子,其中许多为致痛因子,可激活痛觉感受器或诱发痛觉过敏[4]。这些致痛因子包括缓激肽、前列腺素(prostaglandin,PG)E2、大麻素、内皮素、白介素(IL-1β、IL-6、IL-8、IL-15)、集落刺激因子-1(colony-stimulating factor,CSF-1)、趋化因子5(C-C motif chemokine ligand 5,CCL5)、单核细胞趋化蛋白-1(monocyte chemoattractant protein-1,MCP-1)、神经生长因子(nerve grow factor,NGF)、肿瘤坏死因子(tumor necrosis factor,TNF)-α和细胞外ATP等[5]。而在支配骨骼的感觉神经纤维表面有丰富的致痛因子受体和离子通道表达,包括内皮素受体、PG 受体、原肌球蛋白相关激酶A(tropomyosin-related kinase A,TrkA)、缓激肽受体、细胞因子受体、趋化因子受体和嘌呤能受体(P2X3)等[6]。当致痛因子与感觉神经纤维末梢表达的受体及离子通道结合后,可激活痛觉神经元,从而将伤害信号传递到脊髓,然后通过上行系统投射到大脑皮层产生痛觉(图1)。
1.2 肿瘤细胞促进破骨细胞异常活化
生理情况下,骨重塑取决于破骨细胞骨吸收与成骨细胞骨新生之间的平衡。肿瘤骨转移时,此生理性平衡被打破,存在破骨细胞和/或成骨细胞的异常激活[7]。破骨细胞的增殖和分化主要受核因子κB受体活化因子配体(receptor activator for nuclear factor kappa B ligand,RANKL)/RANK通路调节。肿瘤微环境中,肿瘤细胞和T细胞等免疫细胞也会分泌RANKL,另外肿瘤细胞可分泌甲状旁腺激素相关蛋白(parathyroid hormone-related protein,PTHrP)、IL-11等促进成骨细胞表面RANKL表达。升高的RANKL与CSF-1结合后激活其受体RANK从而促进骨髓肿瘤免疫微环境中破骨细胞的分化与成熟。成熟的破骨细胞与骨骼表面紧闭连接形成封闭槽,通过释放大量的H+,进一步激活酒石酸抗酸性磷酸酶(tartrate-resistant acid phosphatase,TRAP)及组织蛋白酶K(cathepsin K,CatK),从而溶解吸收骨骼[8]。吸收槽中H+可作用于骨膜表面感觉神经纤维末梢表达的瞬时受体电位通道1(transient receptor potential channel 1,TRPV1)及酸敏感离子通道3(acid-sensing ion channel 3,ASIC3),从而驱动癌性骨痛的发生及发展(图1)[9]。
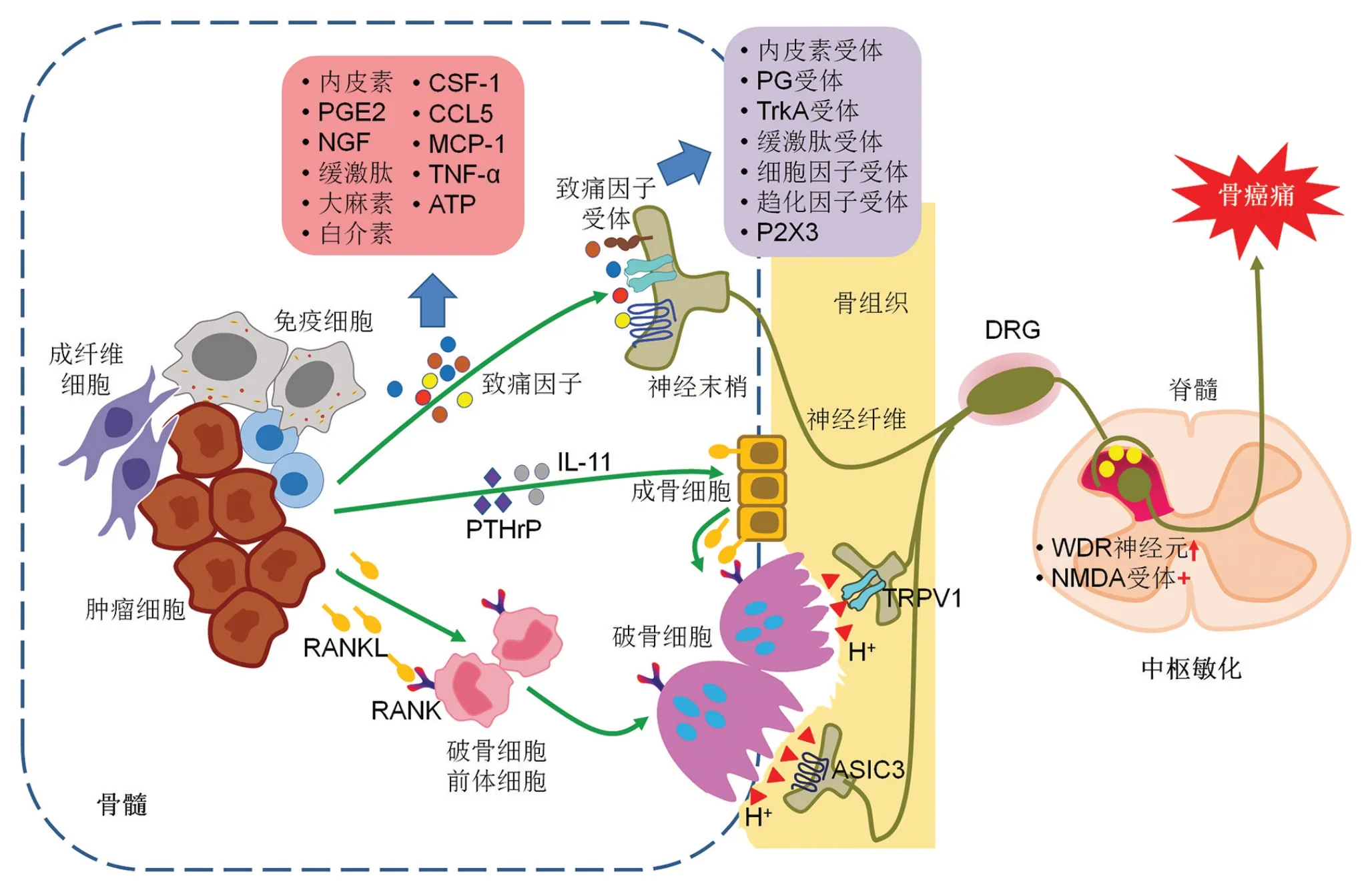
图1 转移性癌性骨痛的分子机制
1.3 中枢敏化
在转移性癌性骨痛中,背根神经节(dorsal root ganglia,DRG)及脊髓背角神经元存在特殊的神经化学性变化[10],提示外周肿瘤与神经纤维末梢相互作用传递疼痛信号进入脊髓后引起中枢敏化,促进癌性疼痛的进展和维持[11]。脊髓切片膜片钳记录结果显示,癌性骨痛动物模型脊髓神经元对诱发的刺激表现出增强的反应,表明神经元的总体兴奋性增强[12]。相较于正常动物,癌性骨痛动物模型中宽动态范围(wide dynamic range,WDR)神经元与伤害感受特异性神经元比例增加,表明脊髓背角浅层神经元的感受野增加,从而导致中枢对传入的低阈值伤害信息作出反应的概率增加,产生中枢敏化[13]。此外,作为中枢性神经递质谷氨酸的受体,谷氨酸的N-甲基-D-天冬氨酸(N-methyl-D-aspartate,NMDA)受体在中枢敏化中也发挥作用。NMDA 受体的持续激活会引发“上发条”(wind-up)现象,即使外周输入信号不变,NMDA 受体也会呈现出4~5 倍过度激活状态,这是中枢敏化的另一重要基础(图1)。
2 癌性骨痛的评估
对于转移性癌性骨痛患者,进行充分的疼痛评估是合理、有效镇痛治疗的前提,应当遵循“常规、量化、全面、动态”的癌痛评估原则[14]。骨痛的评估包括疼痛强度、功能影响及对镇痛药反应等方面[15]。对于认知功能完好的患者,疼痛强度可采用数字评分量表,范围从0(无疼痛)到10(可想象的最严重的疼痛)。此外还可以使用分类量表或视觉模拟量表(无疼痛、轻度疼痛、中度疼痛或重度疼痛)。对认知功能受损的患者,临床医师应注重患者非言语的身体不适表现,如烦躁、表情痛苦、运动功能障碍等,同时观察镇痛药物剂量滴定时患者反应的改善。此外,应重视癌症患者生存质量(qulity of life,QOL)评分中疼痛程度与睡眠精神状态的相互联系,鼓励患者撰写疼痛日记,可为医师判断患者治疗依从性及症状转归提供重要临床线索[15]。
3 转移性癌性骨痛的临床治疗
3.1 药物治疗
3.1.1 非甾体类抗炎药 非甾体类抗炎药(nonsteroidal anti-inflammatory drugs,NSAIDs)基本机制为抑制参与前列腺素生成的环加氧酶(cyclooxygenase,COX)活性,从而产生抗炎、镇痛、解热等作用。动物研究表明,长期应用COX-2 抑制剂也可减轻肿瘤负担,从而进一步缓解癌性骨痛[16]。一项临床荟萃分析表明,尽管NSAIDs 较安慰剂能显著降低癌症相关的疼痛,但其在癌性骨痛中的作用仍有待评价。由于NSAIDs 类药物镇痛效果弱、作用时间短及易引发胃肠道不良反应,其在临床中的治疗作用较为有限[17]。对于轻度疼痛(数字评分量表中评分<4分)患者,建议使用非阿片类镇痛药,如对乙酰氨基酚单独或联合其他NSAIDs[14,18]。
3.1.2 阿片类药物 阿片类药物是癌性骨痛治疗中应用最为广泛的药物之一,给药方式包括口服、经皮、肌肉、静脉及鞘内给药。其主要不良反应包括药物耐受、过度镇静、便秘、恶心呕吐和呼吸抑制[19]。研究表明,长期大量应用吗啡可能促进肿瘤进展,并且是患者不良预后的危险因素[20-21]。因此,临床上应采用基于基因测序的药物基因组学分析,制定个体化治疗方案,可通过使用最小剂量的阿片类药物发挥最佳的止痛效果,同时减少不良反应。
阿片类药物鞘内给药时可联合罗哌卡因或氯胺酮等其他药物,以产生协同效应。鞘内植入镇痛泵时应注意穿刺点局部骨组织肿瘤浸润、鞘内出血及局部感染等禁忌证及风险。由于目前相关临床报道多为非对照研究,因此需要多中心、随机对照临床试验进一步评估阿片类药物通过脊髓途径给药的疗效和安全性[22]。
3.1.3 破骨细胞抑制剂 1)双膦酸盐:双磷酸盐可直接促进破骨细胞凋亡。双磷酸盐与被吸收的骨组织紧密结合并被破骨细胞内吞进入胞内,通过干扰ATP 能量代谢或甲羟戊酸途径引起破骨细胞功能障碍,最终导致破骨细胞凋亡。在肿瘤骨转移过程中,双磷酸盐可改善局部肿瘤骨骼组织的酸性微环境,减少骨破坏及酸敏感离子通道的活化,从而缓解癌性骨痛并降低骨骼相关事件(skeletal-related events,SRE)的风险。其不良反应主要包括肾功能下降及颌骨坏死,临床应用中应密切监测。一项随机临床试验显示,静脉注射4 mg 伊班膦酸可使前列腺癌患者的总体疼痛减轻,与单剂量放疗疗效相当[23]。对于乳腺癌骨转移患者,唑来膦酸每12 周1 次的治疗方案与每4 周1 次治疗方案对于改善患者SRE 的效果并无差异,提示可通过减少唑来膦酸的用量以控制其不良反应[24]。由于在实体瘤(主要为乳腺癌和前列腺癌)以及多发性骨髓瘤引起的骨转移中,双磷酸盐单独短期使用可改善癌性骨痛的证据尚不充分,故临床中双磷酸盐应联合镇痛药物使用[14]。2)地诺单抗:地诺单抗是一种合成的人类单克隆抗体,可与RANKL 结合以阻止其与RANK 相互作用[25]。由于RANKL/RANK信号通路激活是生理和肿瘤病理情况下破骨细胞增殖和分化的必要条件,地诺单抗可抑制破骨细胞分化及功能,从而减少癌性骨痛和肿瘤引起的病理性骨折,改善骨转移患者生存质量。根据欧洲医学肿瘤学会(ESMO)指南,地诺单抗可作为实体瘤和骨髓瘤骨转移时双膦酸盐的替代用药。一项针对乳腺癌骨转移患者的多中心临床试验表明,与唑来膦酸相比,地诺单抗可更为有效地预防并延缓骨痛发展,同时减少阿片类药物的使用量[26]。另一项临床试验中,与双膦酸盐相比,尽管地诺单抗不能减少多发性骨髓瘤患者骨痛程度及时间,但其可降低SRE 风险并促进功能恢复[27]。对于绝经后乳腺癌患者,地诺单抗还可有效预防并减少内分泌辅助治疗所引起的骨质疏松及骨折风险[28]。需指出的是,临床使用地诺单抗时应密切监测颌骨坏死及颚骨坏死等严重并发症[29]。
3.1.4 NGF 阻断剂 由于肿瘤细胞及其基质细胞释放的NGF 可作用于神经纤维末梢TrkA 受体诱发骨痛,并介导神经纤维病理性增生,因此阻断NGF/TrkA信号通路可能缓解癌性骨痛。一项临床前动物研究证实,NGF/TrkA 拮抗剂可减少肿瘤引起的神经出芽或神经瘤的形成[30]。tanezumab是一种重组人源化单克隆抗体,可结合循环及局部组织中的NGF,从而阻止其与TrkA 受体和p75受体相互作用[31]。研究者已在肌肉骨骼慢性非癌性疼痛患者中进行了广泛的tanezumab 相关研究。一项临床试验表明,与安慰剂和NSAIDs类药物相比,tanezumab可有效缓解骨性关节炎痛及下背部疼痛,且无NSAIDs 类药物不良反应[31]。在癌性骨痛领域,两项临床试验前期数据结果显示,tanezumab联合吗啡,可提供吗啡镇痛之外持续的镇痛作用,同时不增加吗啡的不良反应[32]。目前,tanezumab在癌性疼痛治疗中应用的报道较少,尚需要更多临床研究进一步观察及评价。
3.2 非药物治疗
3.2.1 放射疗法 放射疗法(radiotherapy,RT)在转移性癌性骨痛和转移性脊髓压迫(metastatic spinal cord compression,mSCC)治疗中疗效显著[33],被常用于控制癌痛的非药物治疗[15]。多项研究表明,RT 可缓解60%~80%患者的疼痛症状,完全缓解者可达30%[34]。根据WHO 建议,RT 可有效减少镇痛药使用、预防病理性骨折并改善生存质量。目前常用的RT 方法包括外照射放射治疗(external beam radiotherapy,EBRT)、半身照射(half body irradiation,HBI)和放射性药物[35],其中EBRT应用最为广泛且疼痛缓解效果确切。美国放射肿瘤学会(ASTRO)汇总多项随机临床试验发现,EBRT应用不同放疗剂量缓解疼痛时具有等效性[36]。对于转移性癌性骨痛患者,最佳的放射方案为8 Gy单剂量照射[14]。放射性同位素治疗对多部位骨转移引起的癌痛同样具有显著的缓解作用。有研究系统评估了锶、钐或铼对转移性癌性骨痛的治疗效果[37],在短期和中期(1~6 个月)时间内,放射性同位素治疗可在不改变镇痛药物使用方案的情况下,提供更多的疼痛控制,同时不增加白细胞减少和血小板减少等常见不良反应的发生率。在前列腺癌中,一项随机试验评估了镭223对转移性前列腺癌转移性骨痛的治疗效果,发现镭233可显著降低SRE发生率、缓解患者疼痛症状并提高患者生存质量以及生存率,目前镭223为前列腺癌放射性同位素治疗的首选[38]。
3.2.2 神经阻滞或神经损毁 神经阻滞或损毁会导致疼痛传导通路的阻断和破坏,从而抑制信号通过脊髓传递至大脑。阻滞的程度取决于疼痛的严重程度,具体方法包括脊髓或丘脑切开术,以及在神经节上注射神经溶解物质进行微创治疗。与药物治疗相比,介入治疗对耐药及难治性骨痛患者可有效缓解疼痛,且作用时间持久。一项应用神经溶解性交感神经切除术治疗骨盆癌痛的多中心随机对照临床研究表明,早期接受介入治疗患者较晚期接受治疗者的口服镇痛药剂量显著降低,且疼痛控制及生存质量明显改善,提示尽早行神经阻滞治疗可使患者受益增加。同时需要注意,神经破坏及阻滞可引起不可逆性神经损伤,会导致麻木、虚弱、感觉异常、神经认知障碍等并发症[39]。因此应严格选择适应证,权衡风险与收益,并密切监测及治疗可能的并发症。
3.2.3 脊髓电刺激 电神经调节是肿瘤骨转移患者疼痛的第三种非药物治疗方式。该方法包括对周围神经的电刺激以及对脊髓背角和大脑的电刺激。脊髓电刺激最早应用于神经性疼痛的治疗,如蛛网膜炎相关性疼痛,但在感觉伤害性疼痛方面并未得以充分应用。脊髓电刺激可使癌性骨痛患者疼痛程度降低60%左右,从而改善其3年或更长时间的生存质量[40],因此脊髓电刺激在癌痛治疗尤其是难治性癌痛治疗中有广泛的应用前景。
4 结语
综上所述,癌性骨痛的机制复杂,并且会随着癌症的进展而发展。肿瘤骨转移及骨痛可诱发病理性骨折等不良事件,对总体功能和生存质量产生负面影响,并使预后恶化。目前仍缺乏以疼痛为首要观察指标的高质量、多中心、随机对照的临床试验。因此应针对不同的疼痛机制和疾病阶段采用个体化及多模式的治疗策略,以实现最佳的疼痛缓解、改善生存质量并最终延长患者生存期。
