基于随机序列的固有无序蛋白预测算法比较分析
2020-06-15董晴晴赵亚伟于家峰王芳华唐胡成
董晴晴, 赵亚伟, 袁 增, 于家峰, 王芳华, 唐胡成
(1. 德州学院 生物物理研究院 山东省生物物理重点实验室, 德州 253023; 2. 淄博市第四人民医院, 淄博 255067)
近年来,人们发现许多具有重要功能的天然蛋白质在生理条件下并没有稳定三级结构,这类特殊蛋白质通常被称为固有无序蛋白(IDPs,Intrinsically disordered proteins)[1-2]。目前已有大量的研究表明,IDPs普遍存在于自然界中[3-4]。固有无序蛋白根据其结构特征以两种形式存在,一种是完全无序蛋白(IDPs),另一种是序列中交替存在结构有序区和固有无序区(IDRs)的蛋白质。IDPs可以通过与多种分子类型结合、磷酸化等形式获取不同结构来发挥多种生物学功能,因而被认为是蛋白质功能多样化的重要原因[5]。IDPs通常是蛋白质作用网络中的核心蛋白,在包括信号传导、分子识别、细胞周期性调节等各种生命活动中扮演了极为重要的角色,与人类重大疾病密切相关[6-7],因而固有无序蛋白已成为蛋白质科学增长最快的研究领域之一[8]。IDPs的柔性结构特征对传统的“序列→结构→功能”研究模式提出巨大挑战,也为实验上研究该类蛋白提供了科学难题[9-10]。尽管通过NMR、X射线等多种实验手段已经获得了部分IDPs的序列结构数据[11],但与自然界中蛋白质存在的广泛程度和数量相比,对这类蛋白质的认识还相差甚远,人们对此类蛋白的认识还极为有限,其实验研究也存在很大难度,能够实验证实的IDPs数据少之又少。在这种情况下,通过计算方法精准预测IDPs就成为研究此类蛋白质的重要途径[12],也是进一步认识蛋白质功能机制的必要先决条件,对了解相关疾病的致病机制、发现新抑制剂、开发新药均有重要的理论意义和应用价值。然而,尽管近几年研究人员相继提出一批基于不同原理的IDPs预测方法,但预测算法所需训练数据集来源有限,因此预测结果可靠性不高,且不同预测算法之间的结果也具有较为显著的差异[13-15]。因此,如何设计有效方法来对现有IDPs预测算法进行客观评价分析,进而提高IDPs预测效率是IDPs研究的重要工作基础。在此背景下,本文通过人工设计随机蛋白序列作为测试集,对不同类型IDPs预测算法进行系统对比分析,更客观地刻画各算法预测结果差异特征,为今后IDPs研究提供可靠的理论支持。
1 材料与方法
1.1 数据集构建
Disprot数据库[11]是目前IDPs的主要数据来源,该数据库最新版本中提供了800余条经过实验验证的IDPs,已广泛应用于IDPs预测算法的数据集中。此外,许多预测算法还会通过PDB数据库提取IDPs相关数据作为训练集和测试集,然而这些数据集存在明显的倾向性而无法客观刻画IDPs特征[14-15]。本文通过产生随机序列作为独立数据集来完善对不同IDPs预测算法的比较分析,这样可以有效避免所使用数据集已在IDPs预测算法训练集中使用,这些没有刻意的有序区/无序区人为设计偏好的序列可以相对更为客观地展现不同算法的预测结果。从随机角度来讲,有3种常用的产生随机序列的基本策略,一种是各氨基酸在序列中平均使用而排列顺序随机;另一种是完全随机抽取氨基酸,因而各序列中氨基酸组成不同;第3种是基于天然蛋白随机打乱进而得到随机序列。作者近期从二级结构和动力学模拟等角度对多种策略得到的随机序列数据集及由天然蛋白打乱得到的随机数据集进行了研究[16],其结果表明各种随机序列对该论文最终的研究目的影响不大。考虑到第一种策略产生的随机序列中各序列氨基酸百分含量一致,整体上没有任何氨基酸偏好特性,而序列局部区域会有不同的氨基酸使用偏好,会更好地避免数据集设计偏好带来的预测结果偏差,因而本文就以第一种策略得到的随机序列为例进行了研究。通过等比例随机取样的策略从固定的20种氨基酸残基的组合(A、R、N、D、C、Q、E、G、H、I、L、K、M、F、P、S、T、W、Y和V)中随机生成10 000条长度均为60个残基的随机蛋白序列。为了去除数据集中可能的冗余序列,利用CD-HIT程序[17]对随机生成的蛋白序列进行去冗余操作,相似度阈值设为30%,结果显示10 000条随机生成的蛋白序列没有冗余序列存在。
1.2 IDPs预测算法
目前实验研究IDPs难度大,因而针对IDPs的预测算法非常多,但由于缺少公平、可靠的独立IDPs数据集,对这些算法预测效率的有效评价是当前IDPs研究遇到的重要问题。在MobiDB等IDPs数据库中通过采用多种IDPs预测算法来共同判断无序区/有序区,但从结果来看,各算法预测差异很大,最终反而会导致一些真正的无序区被排除掉,保留下来的IDRs区域很少,因此对IDPs预测算法的预测结果进行对比分析来指导IDPs预测算法的合理应用具有重要参考意义。在诸多IDPs预测算法中,IUPred[18]是一款较为经典的基于序列特征的IDPs预测算法,在许多研究中具有广泛应用,而SPINE-D[19]充分考虑了序列保守性特征,在近几年CASP比赛中取得了不错的预测成绩,因而两种算法具有一定的代表性,本文采用了IUPred和SPINE-D算法来完成对比分析工作。其中SPINE-D算法需要调用PSI-BLAST程序对nr(非冗余)数据库进行同源搜索,所以该算法耗时长。
1.3 氨基酸使用偏好分析
我们通过定义AAP值来表示IDPs中各种氨基酸的使用偏好,其计算方法如下:
(1)
(2)

1.4 IDPs预测结果比较
为了比较各种IDPs预测算法预测结果的相似程度,我们定义了公式(3):
(3)
公式(3)中,SI+S、SI和SS分别表示两种算法共同预测得到的无序区氨基酸数目(要具体对应到每个残基位点)、IUPred得到的无序区残基数目和SPINE-D得到的无序区残基数目。显然,0≤K≤1,K=0时表示两种预测算法得到的结果完全不同,K=1时表示两种算法得到的结果完全一致,值越大相符程度越高。
2 结果与讨论
2.1 IDPs预测结果
利用IUPred和SPINE-D程序对10 000条随机蛋白序列分别进行了预测。利用IUPred算法预测时,全部序列均能够返回预测结果;而利用SPINE-D算法时,有7013条序列能够返回预测结果,造成SPINE-D不能预测的原因主要是由于该算法需调用PSI-BLAST程序对nr数据库进行同源搜索,当查询序列在nr数据库中没有同源序列时,就不能生成位置特异性得分矩阵(PSSM),因而无法完成预测。表1中,我们对两种程序的预测结果进行了统计。IUPred预测的10 000条随机序列中,并未发现有无序残基连续长度超过30的序列;然而,SPINE-D有预测结果返回的7013条随机蛋白序列中,总的无序残基个数超过30的有333条(4.75%),无序残基连续长度超过30的有87条(1.24%),并未存在全无序的序列。
图1分析了两种预测软件对每条序列预测得到的无序区碱基数目,其中横坐标表示各随机蛋白序列,纵坐标表示每条序列中预测得到的无序区残基个数。为了便于比较,图1-a中竖线左边的区域是SPINE-D程序能给出预测结果的7013条序列,可见由IUPred得到的预测结果中所有序列的无序区残基个数都在30以下,主要集中在5~12之间;而SPINE-D预测大部分的无序残基数在10~25之间,有333条序列的无序残基数超过30(图1-b)。尽管如此,图1表明随机产生的蛋白序列中有序区要多于无序区。为了进一步说明随机序列中预测得到的无序区残基与天然蛋白中无序区残基的异同情况,对两种程序预测得到的无序区和有序区进行了氨基酸偏好分析(图2)。

表1 IDPs预测结果统计
注:Ⅰ表示统计的随机序列数目;Ⅱ表示两种预测软件成功预测的序列数目;Ⅲ表示预测结果中无序区残基总数大于30个氨基酸的序列数目;Ⅳ表示预测结果中无序区连续长度超过30个氨基酸的序列数目;Ⅴ表示是预测为全无序的序列数目

图1各程序预测结果中无序残基数量分布
图2中对两种预测软件预测得到的有序区和无序区计算了相应的AAP值,通过比较分析,可以发现两种预测方法得到的结果在氨基酸的使用偏好方面是一致的:A、R、N、D、Q、E、G、H、K、P、S和T均偏好出现在无序区域,C、I、L、M、F、W、Y及V均偏好出现在有序区域。在最近工作中[20],作者对基于Disprot数据库构建的一个较大的、实验验证的IDPs数据集进行了深入的有序区/无序区氨基酸使用偏好分析。结果研究表明无序区偏好的氨基酸A、D、E、G、K、P、Q、S及T,有序区偏好的氨基酸是C、F、H、I、L、M、N、R、V、W和Y,这与图2得到的结果是相符的。因此,基于这些随机序列得到的无序区信息与天然蛋白中氨基酸组成相近。

图2 IUPred(a)和SPINE-D(b)预测结果有序区/无序区氨基酸偏好分析
通常情况下,许多文献中将IDPs中序列长度超过30个连续氨基酸的区域称为固有无序区。根据表1的统计结果,SPINE-D程序预测到了87条连续长度超过30个氨基酸的无序区。为此,图3进一步计算了这些连续长度超过30个氨基酸的无序区的氨基酸使用频率。由图3可见,SPINE-D预测结果中A、R、N、D、Q、E、G、H、K、P、S及T的频率均超过5%,而C、I、L、M、F、W、Y和V的使用频率均小于5%。本文所生成的每条随机序列中各氨基酸频率均为5%(1/20),无序区中氨基酸的使用概率若超过5%,则表明该种氨基酸偏好在无序区出现。图3的预测结果与图2是一致的,进一步表明随机序列预测出的有序区/无序区氨基酸使用偏好与天然蛋白是相符的。
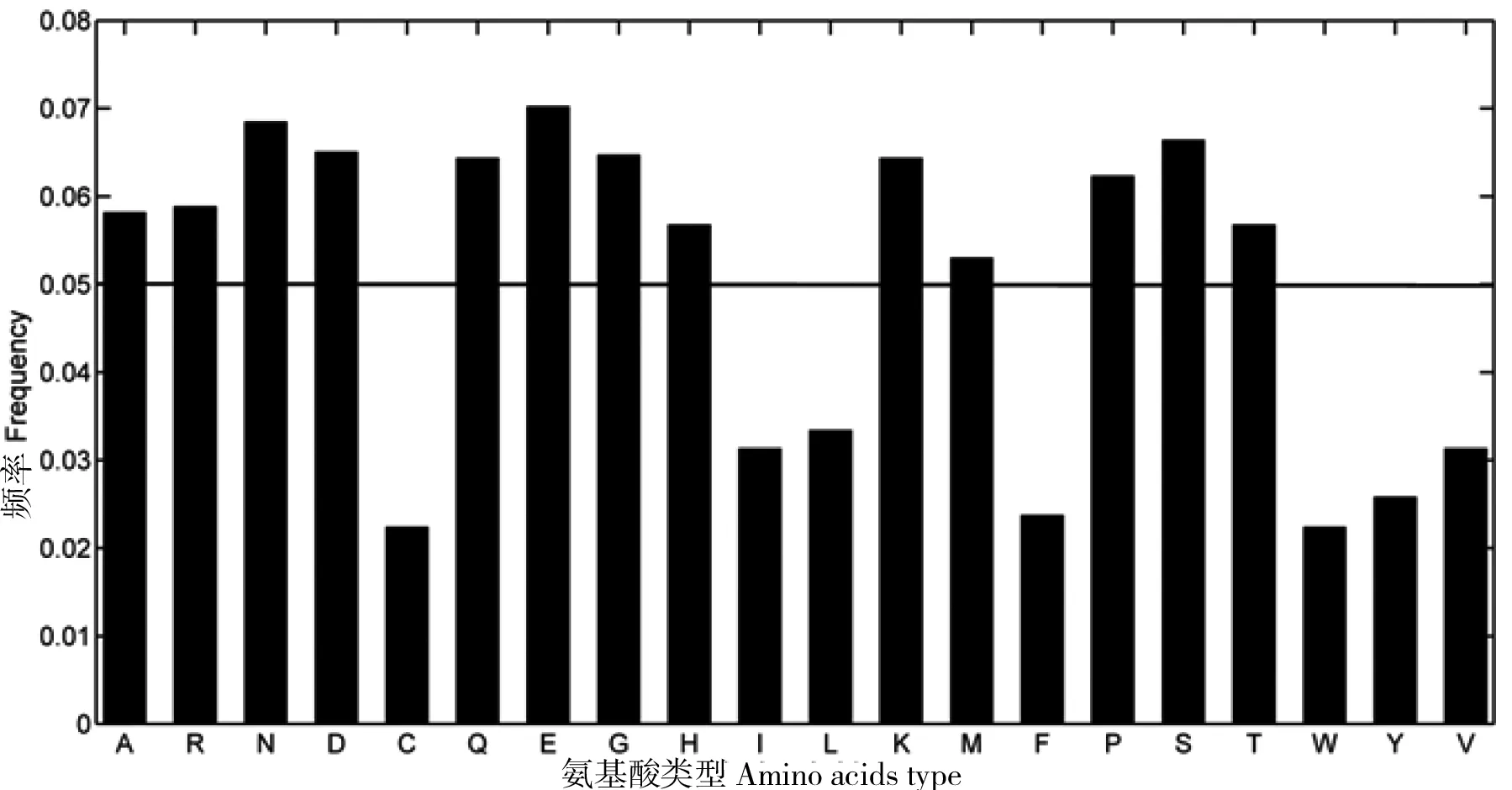
图3 SPINE-D预测结果中连续长度超过30个氨基酸的固有无序区的氨基酸使用频率
2.2 IDPs预测结果比较
上述分析表明两种预测程序得到的具体无序区残基数目具有一定差别。为了进一步分析各预测算法得到的无序区残基在各序列中的位置分布特征,我们将每条序列分为6个区域,每个区域长度为10个氨基酸,各区域无序区残基所占比例表示该区域无序化程度,具体结果见表2。可以看出两种软件预测得到的无序残基主要分布于序列两端,而中间无序残基相对较少。相比之下,SPINE-D预测得到的无序区残基要明显多于IUPred得到的结果。
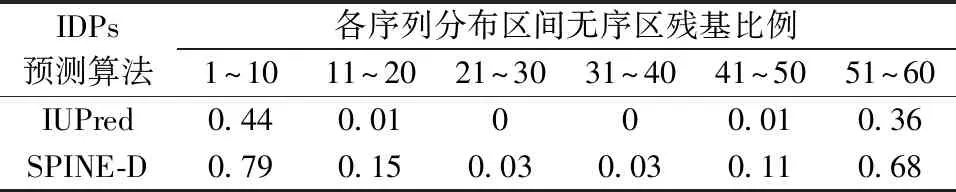
表2 各序列分布区间无序区残基比例
为了更加准确地分析两种IDPS预测软件的预测结果异同特征,图4给出了基于公式3得到的预测结果对比情况。需要说明的是该图分析了两种软件共同预测的7013条序列,横坐标表示K值的分布区间,纵坐标表示各K值分布区间对应的序列数目所占全部序列的百分比。可以看出,两种程序预测相似度对应的K值主要集中在0.65~0.80之间,峰值坐落在0.7附近,而两种预测软件得到的预测结果完全一致的情况很少,这进一步表明IUPred和SPINE-D预测结果具有不同程度区别。
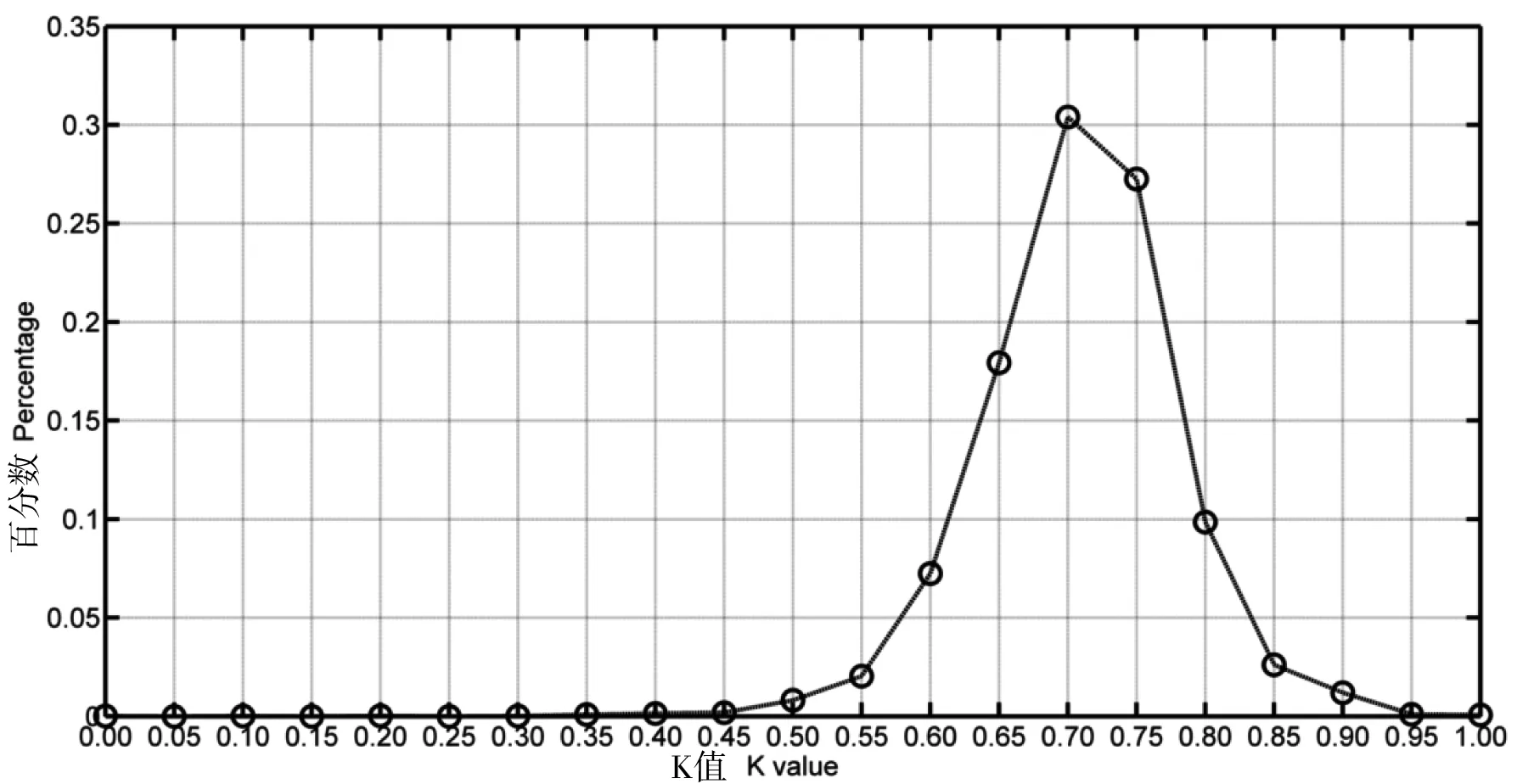
图4 预测结果的相似度情况
3 结论
对IDPs的理论及实验研究还有较大难度,本文基于随机蛋白序列比较系统地对比分析了两种IDPs预测算法的预测结果。预测结果表明,就两种预测算法预测得到的无序残基在序列中位置而言,具有一致性,无序残基大都分布于序列的两端,而具体的对比分析表明这两种预测软件预测结果依然存在较大差异。因此,在今后的实际应用中研究人员还需要进一步整合多种预测算法来评估预测结果的可靠性。
