鸡bmp4过表达和敲除载体构建及活性验证
2020-06-12李婷婷孙长花周舒简张亚妮左其生
李婷婷, 孙长花,2, 周舒简, 金 晶, 张亚妮, 左其生
(1. 扬州大学 江苏省动物遗传繁育与分子设计重点实验室, 扬州 225009; 2. 扬州市职业大学 生物与化工工程学院, 扬州 225009)
PGCs(Primordial germ cells)是所有生殖细胞的祖细胞,在种质资源保护、组织工程治疗等方面均有广泛应用。鸡作为一种重要的模式动物,研究其生殖干细胞PGCs的起源、迁移等机制可为禽类物种保存、良种繁育等提供重要理论参考。研究表明骨形成蛋白4(Bone morphogenetic protein 4,BMP4)参与了哺乳动物胚胎的发育,调节未分化间充质细胞的增殖和分化,在生殖细胞发育过程中,BMP4等信号调节生殖细胞分化为PGCs,启动卵子或精子分化程序,最终形成配子[1-2]。而缺失bmp4的小鼠胚层发育有缺陷,不形成PGCs或尿囊基,最终在胚胎期死亡[3-5]。BMP4是生殖细胞发生分化形成配子的重要信号之一[6],bmp4是BMP4信号通路上的关键基因,研究表明BMP4蛋白在不同物种中的保守性较高[7-8]。
本课题组前期通过添加BMP4蛋白初步建立了鸡PGCs体外诱导模型[9],证明bmp4基因能够调节鸡PGCs的形成,但是否与其在哺乳动物PGCs形成过程中的功能相同仍未知。因此,本研究构建bmp4的过表达及敲除载体,为详细研究bmp4在鸡PGCs形成过程中的功能和机制,分析其在鸡与哺乳动物PGCs形成过程中的差异,完善bmp4调节PGCs形成的调控网络机制提供理论依据。
1 材料与方法
1.1 材料
鸡胚成纤维细胞系DF-1和pCDH-CMV-MCS-EF1-copGFP载体由本实验室保存,LentiCas9-Blast、PGMLV-GM1载体由上海吉满生物技术有限公司提供。PrimeSTAR Max DNA Polymerase、XbaI、EcoR I内切酶购自TaKaRa,DMEM高糖培养基购自Hyclone,胎牛血清购自Gibico,FuGENE®HD Transfection Reagen购自Promega,嘌呤霉素、杀稻瘟菌素、TRIzol、无内毒素质粒小提中量试剂盒、荧光定量试剂盒和基因组提取试剂盒等购自北京天根生物技术有限公司,2×SoSoo同源重组酶、引物合成及测序等由北京擎科生物技术有限公司完成。
1.2 方法
1.2.1bmp4过表达载体构建
基于NCBI数据库中鸡bmp4 CDS区序列(NCBI登录号:NM_205237),利用Primer 5.0软件设计扩增引物,并添加同源臂,F:5′-tgttttgacctccatagaagatTCTAG AATGATTCCTGGTAACCGAATGCTGA-3′;R:5′-cgcggatccgatttaaattcGAATTCAGA GGAAGAGTCCAGCTAGAG CAAGC-3′(小写字母为同源臂),引物由北京擎科生物技术有限公司合成。提取PGCs RNA,反转录成cDNA,以此为模板扩增bmp4 CDS区全长。PCR扩增程序为:98 ℃ 预变性3 min;98 ℃ 变性25 s,59 ℃ 退火30 s,72 ℃ 延伸1 min 20 s, 35个循环; 72 ℃ 再延伸7 min。PCR产物进行琼脂糖凝胶电泳并回收。同时利用限制性内切酶XbaI、EcoR I切割pCDH-MV-MCS-EF1-copGFP空载体,并回收线性载体。利用同源重组法将目的片段连接到线性载体上,连接产物转化感受态细胞,菌液PCR鉴定后阳性菌株提取质粒进行酶切鉴定,鉴定正确的菌株送测序。测序正确后命名为pCDH-CMV-bmp4-EF1-copGFP。
1.2.2 细胞培养与转染
利用0.25%胰蛋白酶消化生长状态良好的DF-1细胞,1600 r/min离心6 min,弃上清。底部细胞用10%FBS-DMEM培养基重悬,以1.5×105个/孔密度铺24孔板,细胞汇合度达70%时进行转染。以pCDH-CMV-bmp4-EF1-copGFP质粒(μg)∶FuGENE(μL)=1∶3比例转染细胞,24 h时观察GFP荧光表达情况,48 h时收集细胞,TRIzol法提取RNA并反转录成cDNA。同时设计定量引物:bmp4-qF:5′-CGCTCTGCCCAAAGCCATGAACT-3′;bmp4-qR:5′-GCACGCTGCTGAGGTTGAAGACG-3′,RT-qPCR检测bmp4的表达变化。
1.2.3bmp4敲除载体构建
根据NCBI上bmp4 CDS区序列,利用Guide Design Resources(https://zlab.bio/guide-design-resources)设计3个sgRNA敲除靶位点:sgRNA1 AAGAAAGTCGCAGAGCTTCA,sgRNA2 CCAAAGCCATGAACTCTTGC,sgRNA3 TGACGGCTGATTTGCTGGGC,分别合成寡聚单链DNA,退火后形成双链。同时设置阴性对照sg-NC,序列为ACGGAGGCTAAGCGTCGCAA。将双链oligo分别连接到CRISPR/Cas9载体(PGMLV-GM1)上,构建重组质粒,分别命名为bmp4-sgRNA1、bmp4-sgRNA2、bmp4-sgRNA3。转化感受态细胞后挑取若干个单菌株测序,阳性菌株提取质粒送上海吉满生物技术有限公司进行慢病毒包裹。
1.2.4bmp4敲除载体活性验证
LentiCas9-Blast载体感染DF-1细胞(MOI=10),并用杀稻瘟菌素(10 μg/mL)持续筛选2周,随后感染sgRNA病毒载体(MOI=10),嘌呤霉素筛选2周后收集细胞提取基因组,T7E1酶切试验检测敲除效率。在bmp4靶位点前后设计引物进行扩增,产物回收后进行T7E1酶切(体系为:10×T7E1 Buffer 1.1 μL,突变型DNA PCR产物400 ng,H2O补足10.5 μL)。酶切样品放入盛有沸水的烧杯中,待其自然冷却至室温,加入0.5 μL T7E1酶,37 ℃酶切30 min后立即加入1 μL DNA Loading Buffer混匀,65 ℃水浴10 min。酶切产物于2%琼脂糖凝胶电泳后观察结果;同时TA克隆试验检测敲除效率,以药物筛选后的细胞基因组为模板扩增bmp4 CDS区,扩增产物连接T载体,转化感受态后每板挑取16株菌落送测序,根据测序结果统计3个载体的敲除效率。
2 结果与分析
2.1 构建bmp4过表达载体
为了构建bmp4过表达载体,本研究根据其CDS区设计特异性扩增引物扩增全长。琼脂糖凝胶电泳在1500 bp附近出现预期结果,最佳扩增温度为59 ℃(图1-A)。在此条件下大量扩增目的条带并连接到双酶切后的线性载体上(图1-B),转化感受态细胞后菌液PCR鉴定和双酶切结果显示插入片段大小正确(图1-C)。测序结果与扩增序列一致,说明bmp4过表达载体pCDH-CMV-bmp4-EF1-copGFP构建成功(图1-D)。
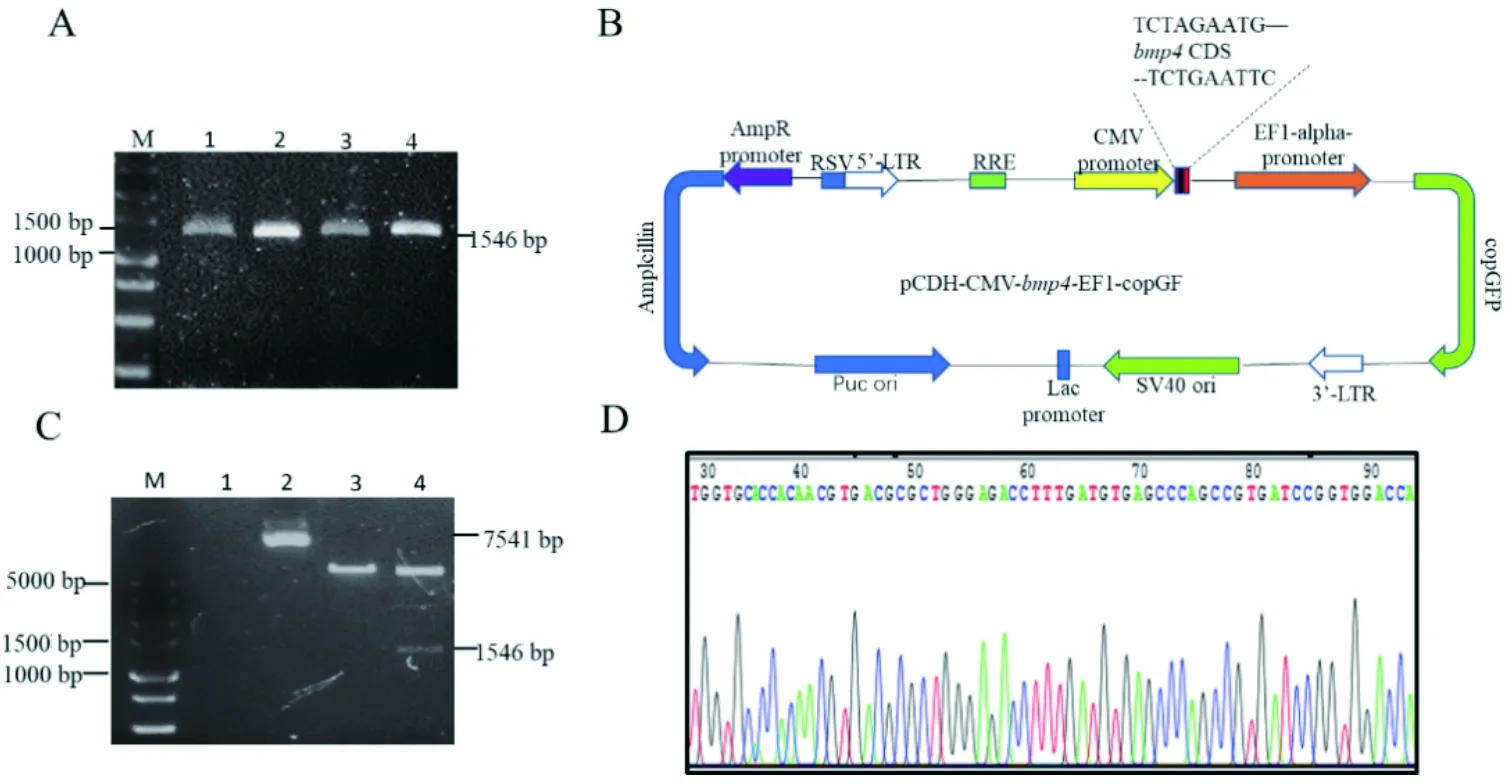
A为bmp4 CDS区,M:DL5000 Marker, 1:60 ℃,2:59 ℃, 3:58 ℃,4:57 ℃; B为pCDH-CMV-bmp4-EF1-copGFP载体;C为bmp4过表达载体双酶切图,M:DL5000 Marker,1:阴性对照(水),2:重组质粒;3:单酶切;4:双酶切;D为bmp4过表达载体测序图
图1bmp4过表达载体构建
Figure 1 Construction ofbmp4 overexpression vector
2.2 bmp4过表达载体在DF-1中的表达
为了验证所构建的pCDH-CMV-bmp4-EF1-copGFP过表达载体是否能表达GFP蛋白,用测序正确的菌株提取质粒转染DF-1细胞,48 h时观察到绿色荧光(图2-A),说明转染成功。收集细胞提RNA,RT-qPCR检测结果显示,转染过表达载体后,bmp4表达量显著高于转染过表达空载体及空白对照组(P<0.01),且空载与空白对照无显著差异(图2-B)。以上结果均说明构建的bmp4过表达载体有活性,可启动bmp4的表达。
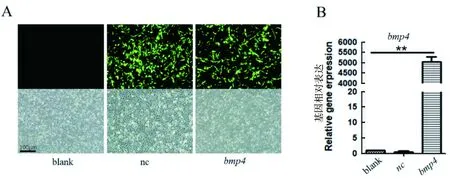
A:bmp4过表达载体转染DF-1细胞(标尺100 μm); B:定量检测bmp4的表达(**表示P<0.01)
图2bmp4过表达载体活性验证
Figure 2 Validation ofbmp4 overexpression vector activity
2.3 靶向bmp4的sgRNA载体构建
为了构建bmp4敲除载体,利用Guide Design Resources(https://zlab.bio/guide-design-resources)在bmp4 CDS区设计3个sgRNA敲除靶点,构建bmp4敲除载体bmp4-sgRNA1、bmp4-sgRNA2和bmp4-sgRNA3(图3-A),测序结果显示载体构建成功(图3-B),送上海吉满生物技术有限公司进行慢病毒包裹,载体滴度分别为1.16×109、5.23×108和1.07×109TU/mL。
2.4 bmp4敲除载体活性验证
为了验证3个sgRNA载体的敲除活性,将其转染DF-1细胞,杀稻瘟菌素和嘌呤霉素各筛选2周(图4-A),收集细胞提取基因组做T7E1酶切检测。酶切产物进行2%琼脂糖凝胶电泳,敲除组有两条带,野生型和空载组仅有一条带(图4-B),说明3个sgRNA载体对bmp4均有敲除作用。同时,TA克隆测序结果进一步说明3个sgRNA载体对bmp4有敲除作用(图4-D),效率分别为6.25%、12.5%和18.75%,可见bmp4-sg3RNA敲除效率最高(图4-C),可用于后续试验。
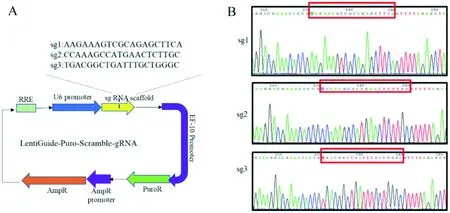
A:bmp4-sgRNA重组载体; B:从上到下分别为敲除载体sg1、sg2及sg3测序峰图
图3bmp4敲除载体构建
Figure 3 Construction ofbmp4 knockout vector
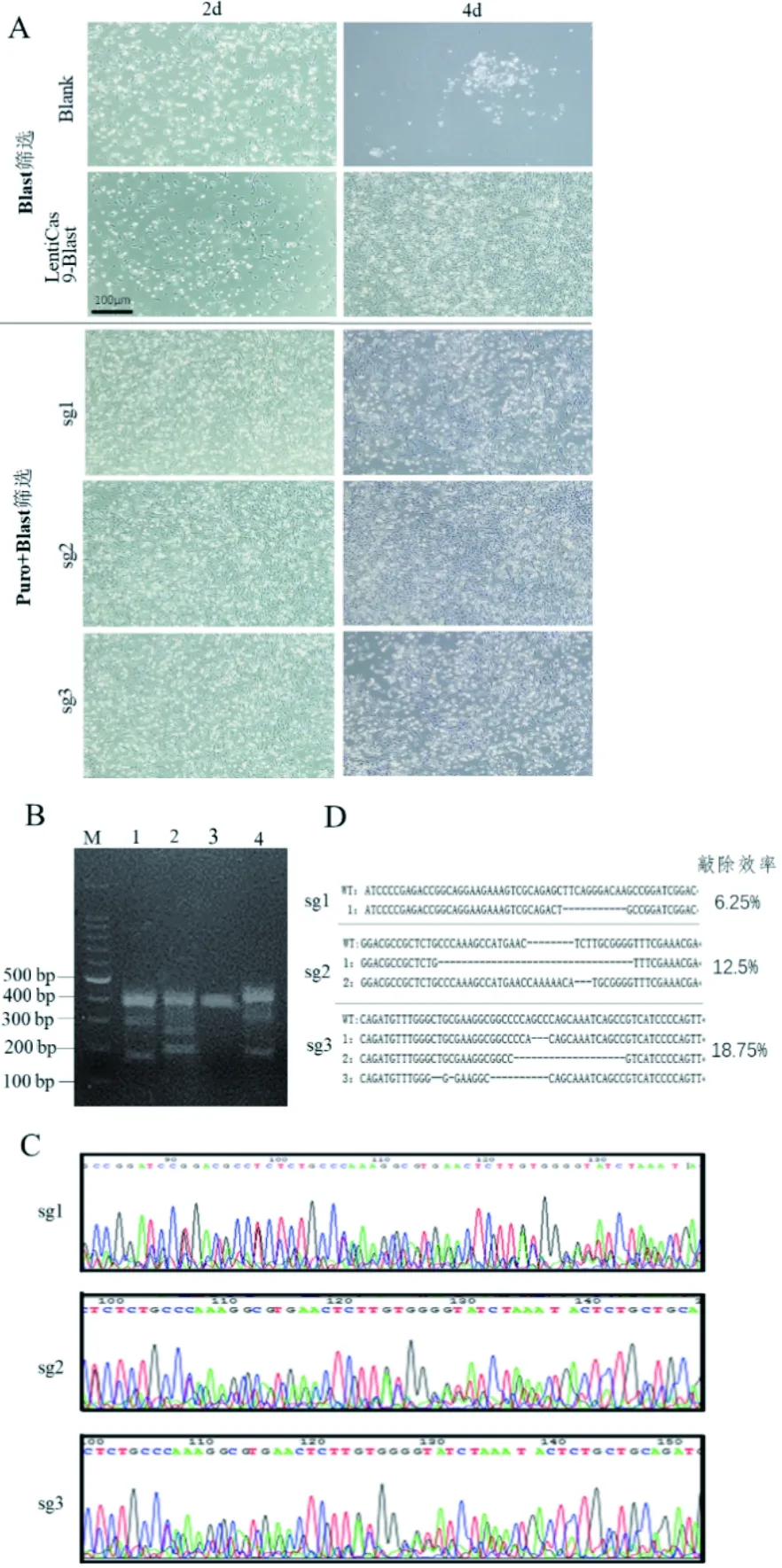
A:敲除载体转染DF-1药物筛选后细胞形态图(标尺100 μm);B:T7E1酶切图, M: 100 bp DNA Ladder, 1:sgRNA1, 2: sgRNA2, 3:野生型4:sgRNA3; C:从上到下分别为sg1、sg2、sg3载体TA克隆测序峰图;D:sg1、sg2、sg3载体敲除结果及效率统计
图4bmp4敲除载体体外活性验证
Figure 4 The activity verification ofbmp4 knockout vectorinvitro
3 讨论
Bmp4调节哺乳动物原始生殖细胞形成的研究已较为清楚,通过与靶细胞上的受体结合激活细胞内SMADs信号转导蛋白,将信号传入细胞核进而调节PGCs的形成[10-11],但其在鸡PGCs形成中的作用仍不明确。目前市场上已有商品化的BMP4蛋白及其抑制剂,抑制剂主要是抑制BMP的I型受体(ALK2、ALK3、ALK6)及阻碍SMAD1/5/8的磷酸化,阻断的效果与使用量有关[12],用于体内试验时均有一定局限性,且不能直接作用于bmp4。因此,本实验构建了bmp4的过表达载体,转染DF-1细胞后bmp4表达量显著升高。同时构建了bmp4的敲除载体,特异性靶向bmp4,在基因组上实现bmp4的敲除,彻底抑制其表达,阻断bmp4的作用,便于bmp4功能及机制的探索。但敲除效率较低,使得转染后阳性细胞的筛选和检测工作较为困难。分析其原因,sgRNA和Cas9蛋白位于两个载体上,需先转染Cas9蛋白载体,杀稻瘟菌素筛选后再转染sgRNA载体,较难保证细胞内同时转入了两个载体。而且,经过药物筛选后细胞状态变差,增加了sgRNA转染的难度,导致本实验最终敲除效率偏低。同时本实验进行T7E1酶切和TA克隆检测前仅进行药物筛选未挑取单细胞株,可能混杂有部分bmp4未敲除的细胞,导致结果偏低。但基因组上已检测到bmp4有敲除,因此,本研究中bmp4过表达及敲除载体构建成功,可用于后续bmp4的功能及机制研究。
相对于利用RNA干扰技术及使用抑制剂降低基因的表达水平以研究基因功能而言,在基因组上进行敲除能更彻底地阻断基因的表达,更准确地研究其功能。现有的基因敲除技术主要有锌指核酸酶(ZFNs)、转录激活样效应因子核酸酶(TALEN)和成簇规律间隔短回文重复(CRISPR / Cas9)系统,其中CRISPR / Cas9技术相比于前两者载体构建简单、工作量较小、成本低,且敲除效率高,已快速广泛地应用于动、植物基因编辑方面的研究[13-14]。近几年利用CRISPR/Cas9技术对PGCs进行基因编辑的研究不断深入[15-19],已有文献报道CRISPR/Cas9技术可应用于家禽研究领域进行基因编辑[20-22]。但CRISPR/Cas9技术也存在一些缺点,如需要Cas9蛋白和sgRNA同时表达、脱靶效率较高等。现在CRISPR/Cas9敲除体系主要有Cas9蛋白和sgRNA位于同一个载体上及分别构建到两个载体上两种形式。本实验中Cas9蛋白和sgRNA分别在两个载体上,降低了单质粒表达系统脱靶率较高的问题。本研究在后续实验中将继续优化载体及转染条件,提高敲除效率,为后期bmp4功能验证及具体分子机制解析奠定基础。
