基于线粒体Cytb和D-Loop序列的大獭蛤5个地理群体遗传多样性研究
2020-06-10王伟峰陈秀荔朱威霖王焕岭
陈 康,王伟峰,陈秀荔,朱威霖,王焕岭
(1.华中农业大学水产学院,湖北武汉 430070;2.广西壮族自治区水产科学研究院,广西水产遗传育种与健康养殖重点实验室,广西南宁 530021)
大獭蛤(Lutrariamaxima)隶属双壳纲(Bivalvia),异齿亚纲(Heterodonta),真瓣鳃目(Eulamellibranchia),蛤蜊科(Mactridae),獭蛤属,为营埋栖生活的二倍体贝类,广泛分布于中国中南部沿海,北至福建省,南至海南省,尤其在北部湾地区较为丰富,目前通过人工养殖,已发展成为广西、广东等地浅海水产养殖的主要品种之一[1-3]。近年来,我国沿海地区建设了众多高能耗、高排放的大型项目,大量生活污水和工业废水排入海中,导致海水污染加重,赤潮暴发次数增加,水生生物栖息环境被严重破坏[4-5]。据相关报道,在北部湾贝类市场检测的所有贝类生物中均含有麻痹性贝类毒素[6],以贝类为主的经济物种面临巨大的生存挑战和繁殖压力。另一方面,人工育种技术不断成熟[7],育种范围相对集中,导致大獭蛤近亲繁育加剧,很可能造成种质资源不断退化。因此,对大獭蛤野生群体的资源评估显得十分必要。
为了促进大獭蛤资源的可持续利用,本研究采用分析遗传多样性的方式为其遗传育种和种质资源的评估提供重要的依据[8]。由于多态性信息丰富、遗传稳定性强和检测手段相对简便等特点,DNA分子标记在群体遗传结构及其分化研究方面具有无可比拟的优势[9]。其中线粒体DNA(mitochondrial DNA,mtDNA)是闭合、环状的双链DNA分子,其作为遗传信息的重要载体,具有母系遗传、结构简单、进化速度快、几乎无重组及不同的区域进化速度存在差异等特点,使得mtDNA成为一种重要的分子标记[10]。在mtDNA中,细胞色素b(Cytochrome b,Cytb)和mtDNA控制区(又称D-环区,control region displacement loop,D-Loop) 的进化速度存在差异且变异速率快[11-12]。因此,利用Cytb和D-loop序列分析和对比不同群体mtDNA多态性,能够揭示种内或种间的亲缘关系[13]。近年来,通过分析Cytb和D-Loop 序列的碱基信息检测水生生物遗传多样性已被广泛应用,如基于Cytb基因序列发现中国养殖区和日本原产地的虾夷扇贝(Patinopectenyessoensis)出现明显的遗传分化[14];王剑平等[15]基于Cytb基因发现洞庭湖河蚬(Corbiculafluminea)遗传分化弱,变异主要来自群体内;GUO等[16]利用Cytb和D-Loop序列得出西藏雅鲁藏布江6个地理群体的裸裂尻鱼(Schizopygopsisyounghusbandi)的遗传多样性水平较低,并且遗传变异来自群体内部,这些研究为水生生物种质资源的开发与利用奠定了理论依据。
目前,关于大獭蛤的研究主要集中在营养成分分析与评价[17]、繁殖特性[18]及人工育苗[7,19]等方面。而关于大獭蛤种质资源评估的研究鲜有报道,相关研究仅见李斌等[20]采用形态学特征和RAPD技术,对3个地理群体的施氏獭蛤(L.siebaldii)与越南大獭蛤(L.maxima)遗传差异进行了比较分析。因此为了评估我国不同地理群体大獭蛤资源,本研究利用Cytb和D-Loop序列,对我国大獭蛤5个地理群体的遗传多样性、群体结构和群体历史动态进行分析,以期揭示大獭蛤种质资源的遗传背景,为大獭蛤的人工繁育和资源保护提供理论依据。
1 材料与方法
1.1 实验材料与DNA提取
大獭蛤分别采自广西北海(BH)、广西涠洲岛(WZD)、广东湛江(ZJ)、福建厦门(XM)和福建福州(FZ)5个地区(图1),取前后闭壳肌组织保存于95%乙醇中,用于基因组DNA的提取,每个群体的个体数及遗传多样性参数见表1。采用醋酸铵法[21]从肌肉组织中提取大獭蛤基因组DNA,1%琼脂糖凝胶电泳和Nanodrop测定DNA的质量和浓度。
1.2 PCR扩增
根据NCBI数据库中大獭蛤mtDNA全序列(Genbank:NC_036766.1)利用Primer Premier 5.0设计引物用于扩增Cytb和D-Loop序列。引物序列分别为,CytbF:5′ -TGCGGCTGTCTGGTATTGA-3′、CytbR:5′-AACCCTTTCATCGTCCCACTA -3′和D-Loop F:5′-ATTAGAATACGCCGTTGAAG-3′、D-Loop R:5′-GAGTAGTTACATCCTGCTTAC-3′,由武汉天一辉远生物科技有限公司合成。
PCR总反应体积为10 μL,反应体系为:10×PCR buffer 1 μL,dNTPs 0.4 μL,双蒸水6.6 μL,DNA模板1 μL,ESTaqDNA聚合酶0.2 μL,左右引物各0.4 μL。扩增程序为:预变性 94℃ 3 min,变性94℃ 30 s,退火30 s(D-Loop为52℃,Cytb为54℃),延伸72 ℃(D-Loop为45 s,Cytb为105 s),共32个循环,终延伸72℃ 7 min。扩增产物经1%琼脂糖凝胶电泳检测后,选择目的条带明显且特异性好的PCR产物送武汉天一辉远生物科技有限公司测序。
1.3 数据分析
基于测序峰图使用DNAstar软件包[22]中的Seqman软件对测序结果进行分析校对,以确认序列质量,并与NCBI数据库中公布的大獭蛤mtDNA进行对比,获得不同大獭蛤群体的Cytb和D-Loop序列。MEGA7.0[23]计算Cytb和D-Loop序列的碱基组成并使用邻接法(neighbor-joining,NJ)构建系统发育树,采用Bootstrap(重复次数1 000)检验聚类树各分支置信度。DNAsp5[24]获得Cytb和D-Loop序列的核苷酸多样性指数(nucleotide diversity,Pi)、平均核苷酸差异数(average number of nucleotide differences,K)、单倍型数(number of haplotypes,H)、单倍型多样性指数(haplotype diversity,Hd)及多态位点数(number of polymorphic sites,S)。运用Arlequin3[25]计算群体间的遗传分化系数和扩张参数τ,并结合中性检验和核苷酸不配对分布曲线来推测大獭蛤种群的历史动态情况;根据公式τ= 2ut和T=t×(代时),估算种群扩张时间T[26]。其中u=μk,μ为研究序列变异速率,k为研究的序列长度(bp);大獭蛤性成熟约为1年,则代时为1[18]。本研究中,Cytb和D-loop的变异速度分别为(1.0%~2.5%)/百万年[27]、(3%~10%)/百万年[28]。

图1 大獭蛤采样分布图 Fig.1 Sampling sites for L. maxima注:BH、WZD、ZJ、XM、FZ分别代表广西北海、广西涠洲岛、广东湛江、福建厦门、福建福州;●代表采样点的位置Note:BH, WZD, ZJ, XM, FZ represent Beihai of Guangxi, Weizhou Island of Guangxi, Zhanjiang of Guangdong, Xiamen and Fuzhou of Fujian respectively;●indicates sampling site
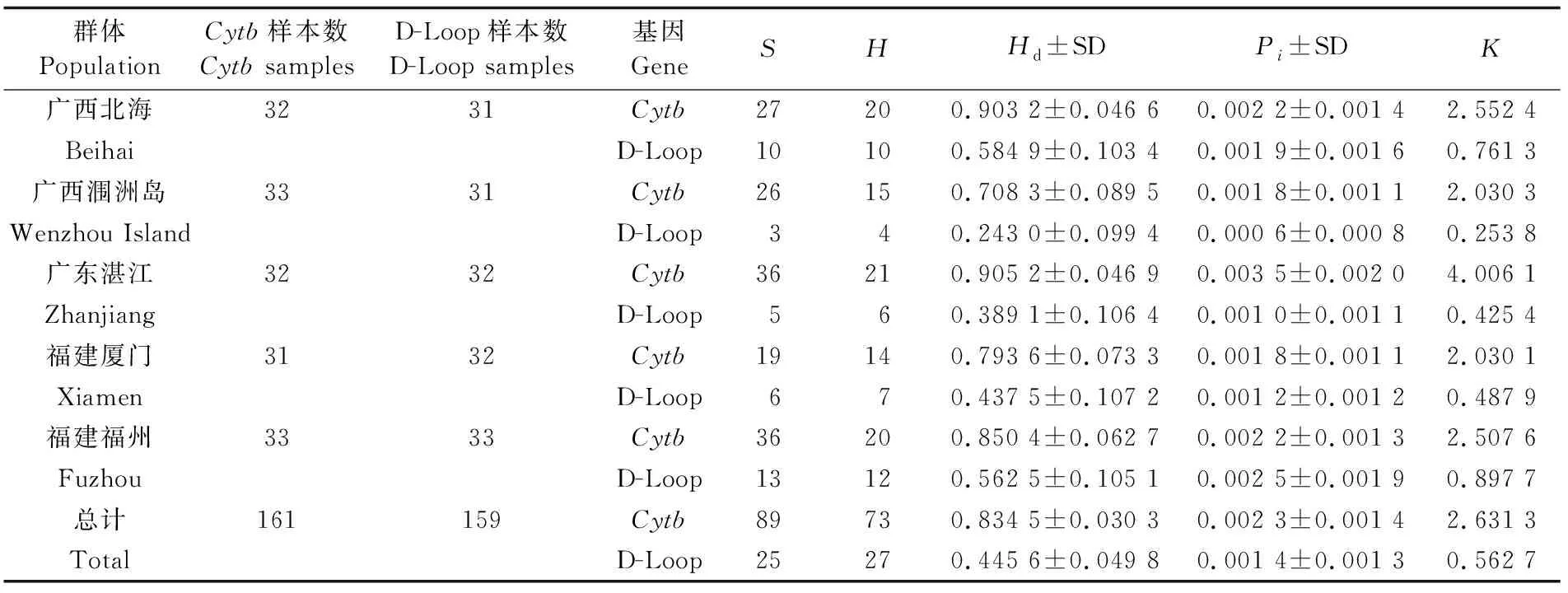
表1 大獭蛤5个群体的遗传多样性参数Tab.1 Genetic diversity parameters of five populations of L. maxima
注:S:多态位点数;H:单倍型数;Hd:单倍型多样性指数;Pi:核苷酸多样性指数;K:平均核苷酸差异数
Note:S: number of polymorphic sites;H: number of haplotypes;Hd: haplotype diversity;Pi: nucleotide diversity;K: average number of nucleotide differences
2 结果与分析
2.1 大獭蛤线粒体Cytb和D-Loop的序列特征和遗传多样性
测序结果经软件处理后分别获得了大獭蛤Cytb基因序列(1 155 bp)和D-Loop序列(412 bp)。利用MEGA7软件计算序列的碱基组成,结果显示5个群体间Cytb和D-Loop序列的碱基含量基本一致,Cytb基因和D-Loop序列4种碱基T、C、A、G的平均含量分别占其全长的43.60%、13.53%、20.61%、22.26%和23.31%、25.25%、39.29%、12.16%,且两序列的A+T含量明显高于G+C含量(表2),表现出了AT偏移,这与大多数水生动物的已知模式一致。使用DNAsp5对大獭蛤5个地理群体的遗传参数进行计算,结果显示Cytb基因在5个群体的161个个体的核苷酸多样性指数为0.002 3±0.001 4,单倍型多样性为0.834 5±0.030 3,平均核苷酸差异数为2.631 3,共检索到89个多态位点和73种单倍型。D-Loop序列在5个群体中159个个体的核苷酸多样性指数为0.001 4±0.001 3,单倍型多样性为0.445 6±0.049 8,平均核苷酸差异为0.562 7,共发现25个多态位点和27种单倍型。其中,北海群体的单倍型多样性指数Cytb序列为0.903 2,D-Loop序列为0.584 9,该群体的单倍型也是比较丰富的(表1)。
2.2 大獭蛤种群的遗传分化分析
在种群遗传学中,遗传分化指数(Fst)可反映群体间的遗传分化程度,有学者将遗传分化程度分为弱分化(Fst<0.05)、中度分化(0.05≤Fst≤0.25)、高度分化(Fst>0.25)3个等级[29]。本研究基于Cytb和D-Loop序列分析,北海、福州、涠洲岛、厦门、湛江5个群体Fst分别在-0.008 0~0.010 1(P>0.05)和-0.014 3~-0.007 2(P>0.05)之间变动,同时5个地理群体间的遗传距离分别在0.001 8~0.002 9和0.000 8~0.002 0之间(表3,表4)。结果表明,每个群体之间遗传分化程度都较弱(Fst<0.05),遗传距离较近,说明5个群体之间具有较高的遗传同质性。
基于Cytb对大獭蛤群体AMOVA分析表明:99.68%的差异属于群体内差异,0.32%为群体间差异(表5);基于D-Loop对大獭蛤群体AMOVA分析表明:101.16%的差异属于群体内差异,-1.16%为群体间差异(表5),表明群体内遗传变异远远高于群体间的变异。
2.3 系统进化树
基于Cytb基因(图2-A)和D-Loop序列(图2-B)的两个系统发育树都分为两大支系,未形成明显的单系群,5个地理群体间相互交叉,无明显的地理群体聚类,与上述群体间遗传距离弱、群体间分化指数Fst值低的结果相符。
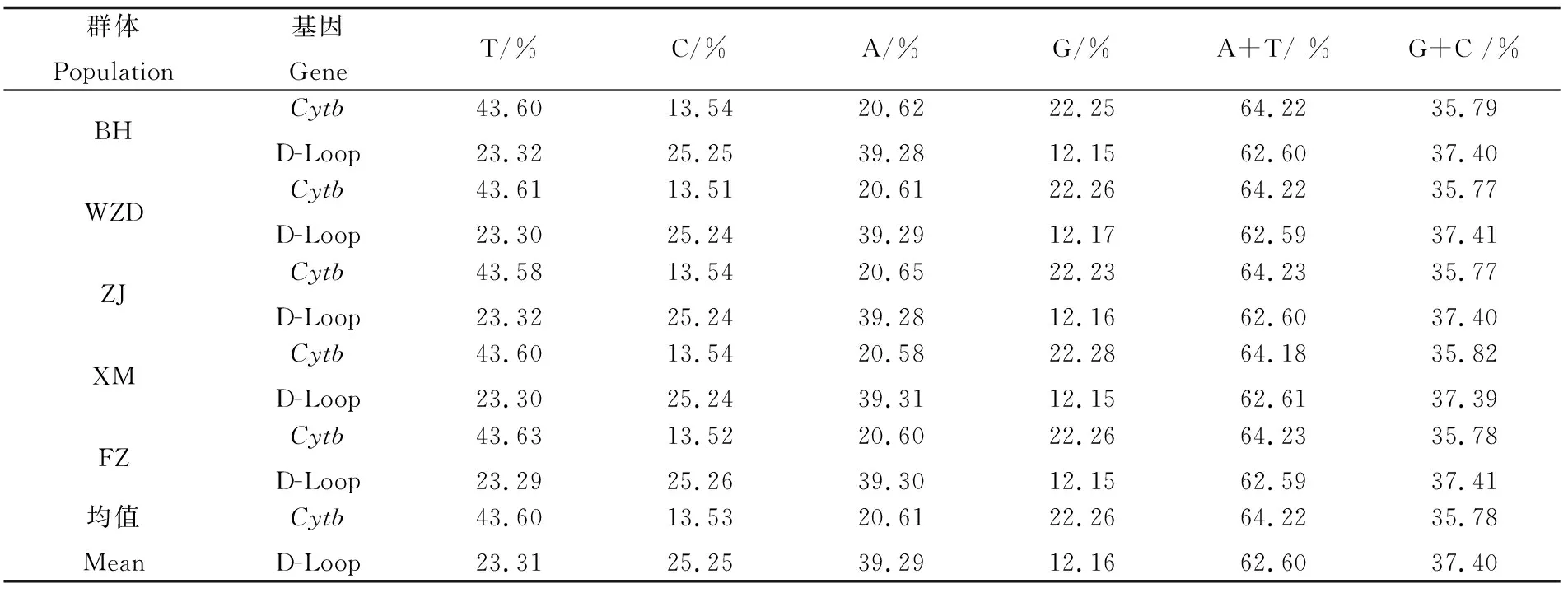
表2 大獭蛤5个群体Cytb和D-Loop序列的碱基组成Tab.2 Base composition of Cytb and D-Loop sequences in five populations of L. maxima
注:BH、WZD、ZJ、XM、FZ分别代表广西北海、广西涠洲岛、广东湛江、福建厦门、福建福州
Note: BH, WZD, ZJ, XM, FZ represent Beihai of Guangxi, Weizhou Island of Guangxi, Zhanjiang of Guangdong, Xiamen and Fuzhou of Fujian respectively

表3 基于Cytb基因的大獭蛤群体间遗传距离及遗传分化指数FstTab.3 Genetic distance and fixation index (Fst) between every two populations of L. maxima based on Cytb gene
注:对角线下数据为群体间遗传距离,对角线上数据为遗传分化指数Fst;BH、WZD、ZJ、XM、FZ分别代表广西北海、广西涠洲岛、广东湛江、福建厦门、福建福州;Fst值均无显著性(P>0.05)
Note:Data below the diagonal mean genetic distance, above the diagonal mean fixation indexFst; BH, WZD, ZJ, XM, FZ represent Beihai of Guangxi, Weizhou Island of Guangxi, Zhanjiang of Guangdong, Xiamen and Fuzhou of Fujian respectively; allFstvalues were not significant (P>0.05)

表4 基于D-Loop序列的大獭蛤群体间遗传距离及遗传分化指数FstTab.4 Genetic distance and fixation index (Fst) between every two populations of L. maxima based on D-Loop sequence
注:对角线下数据为群体间遗传距离,对角线上数据为遗传分化指数Fst;BH、WZD、ZJ、XM、FZ分别代表广西北海、广西涠洲岛、广东湛江、福建厦门、福建福州;Fst值均无显著性(P>0.05)
Note:Data below the diagonal mean genetic distance, above the diagonal mean fixation indexFst; BH, WZD, ZJ, XM, FZ represent Beihai of Guangxi, Weizhou Island of Guangxi, Zhanjiang of Guangdong, Xiamen and Fuzhou of Fujian respectively; allFstvalues were not significant (P>0.05)

表5 大獭蛤5个地理群体Cytb和D-Loop序列的AMOVA结果Tab.5 AMOVA result of five populations based on Cytb and D-Loop sequences
注: Va、Vb 分别表示群体间变异和群体内变异
Note: Va, Vb mean variations among populations and within populations
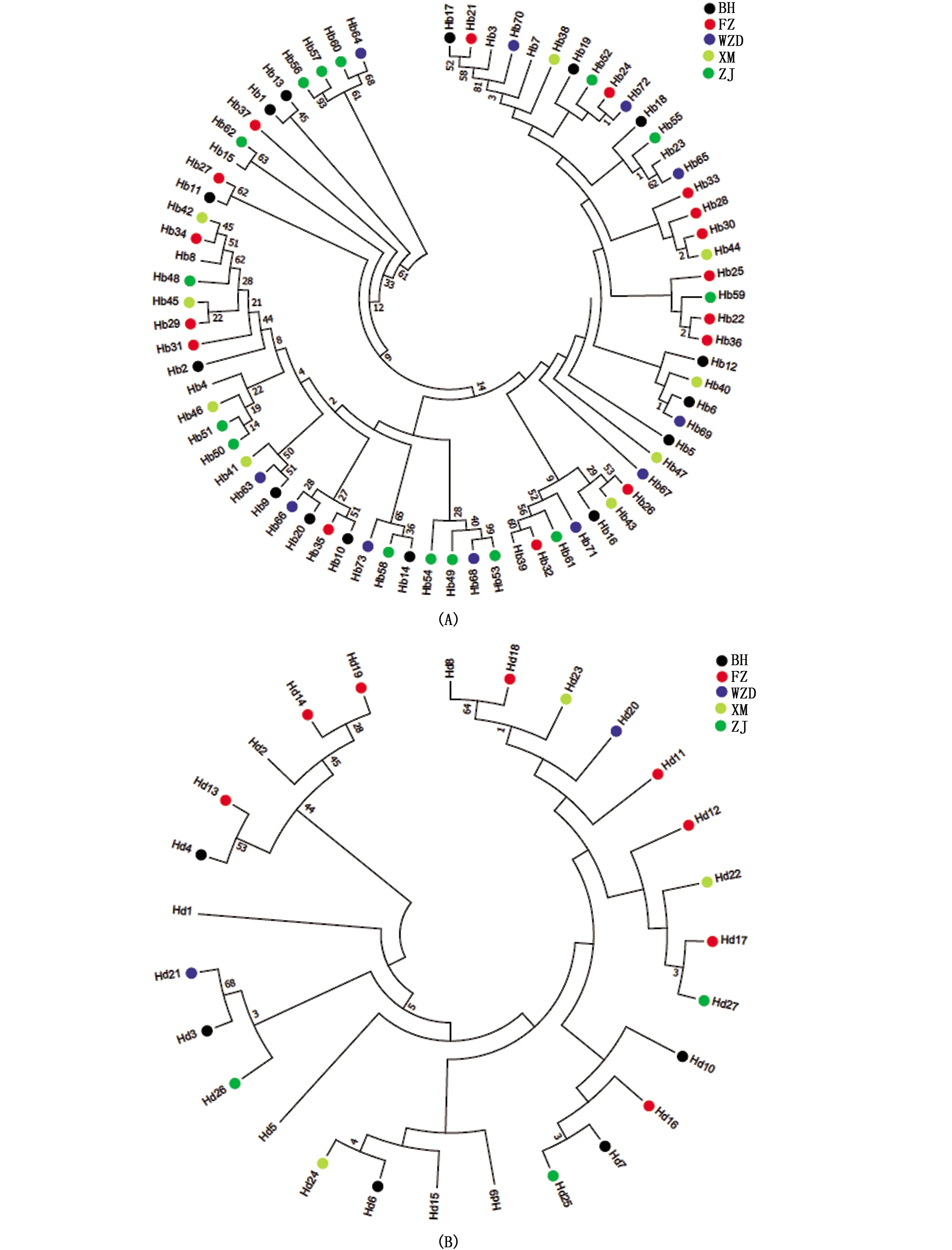
图2 基于Cytb(A)基因和D-Loop(B)序列构建大獭蛤单倍型NJ树Fig.2 The NJ phylogenetic tree of L. maxima based on Cytb (A) and D-Loop(B) haplotypes注:每个群体的私有单倍型都用圆圈标记,而群体之间的其他共享单倍型则不标记。BH、WZD、ZJ、XM、FZ分别代表广西北海、广西涠洲岛、广东湛江、福建厦门、福建福州Note:The private haplotypes of each population are marked with circles, and the other shared haplotypes among populations are not marked. BH, WZD, ZJ, XM, FZ represent Beihai of Guangxi, Weizhou Island of Guangxi, Zhanjiang of Guangdong, Xiamen and Fuzhou of Fujian respectively
2.4 种群动态
由于大獭蛤5个地理群体间的遗传分化程度较弱,故分析其历史动态信息时将其看作一个整体,并依据Tajima’sD[30]和 Fu’sFS[31]值中性检验和核苷酸不配对分布曲线来判定大獭蛤在过去是否发生了种群历史扩张。大獭蛤Cytb和D-Loop序列的中性检验分析结果如表6所示。基于Cytb序列各群体的Tajima’D和Fu’sFS,均为负值,统计学分析都存在显著差异(P<0.05),将5个群体作为一个整体进行中性检验,其Tajima’sD和 Fu’sFS值分别为-2.229 0(P<0.01)和-12.658 8(P<0.01),Cytb序列核苷酸错配分布图为单峰(图3-A)。基于D-Loop序列各群体的Tajima’D和Fu’sFS,均为负值,统计学分析都存在显著差异(P<0.05),将5个群体作为一个整体进行中性检验,其Tajima’sD和 Fu’sFS值分别为-1.943 2(P<0.05)和-6.248 6(P<0.05),D-Loop序列核苷酸错配分布图为单峰(图3-B)。这些结果表明大獭蛤在历史上发生过明显的种群扩张。在拟合度检验中(表6),基于Cytb和D-Loop序列分析,5个群体的偏离方差(sum of squared deviation)和Raggedness统计量(R)的值都较小,且大部分不存在显著差异(P>0.05)。从整体来看,这两个参数的值都较小,均未达到显著水平(P>0.05),同样表明大獭蛤历史上经历了种群扩张。基于Cytb和D-Loop序列的平均扩张参数τ值分别为1.523 8、1.038 7,推测出种群扩张时间距今约为1.3×104~6.6×104年,表明大獭蛤种群扩张时间约在更新世晚期。
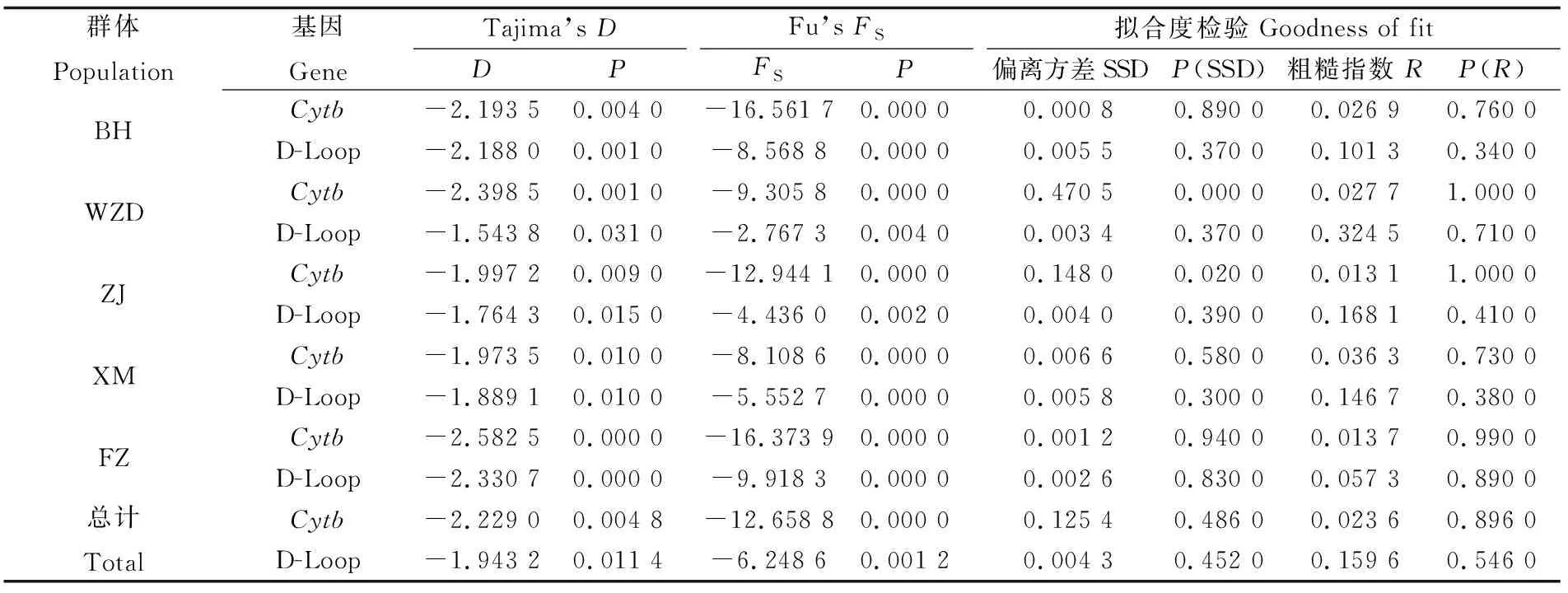
表6 大獭蛤5个群体的中性检验、拟合度检验Tab.6 Neutrality test and test of goodness of fit in five populations of L. maxima
注:BH、WZD、ZJ、XM、FZ分别代表广西北海、广西涠洲岛、广东湛江、福建厦门、福建福州
Note:BH, WZD, ZJ, XM, FZ represent Beihai of Guangxi, Weizhou Island of Guangxi, Zhanjiang of Guangdong, Xiamen and Fuzhou of Fujian respectively
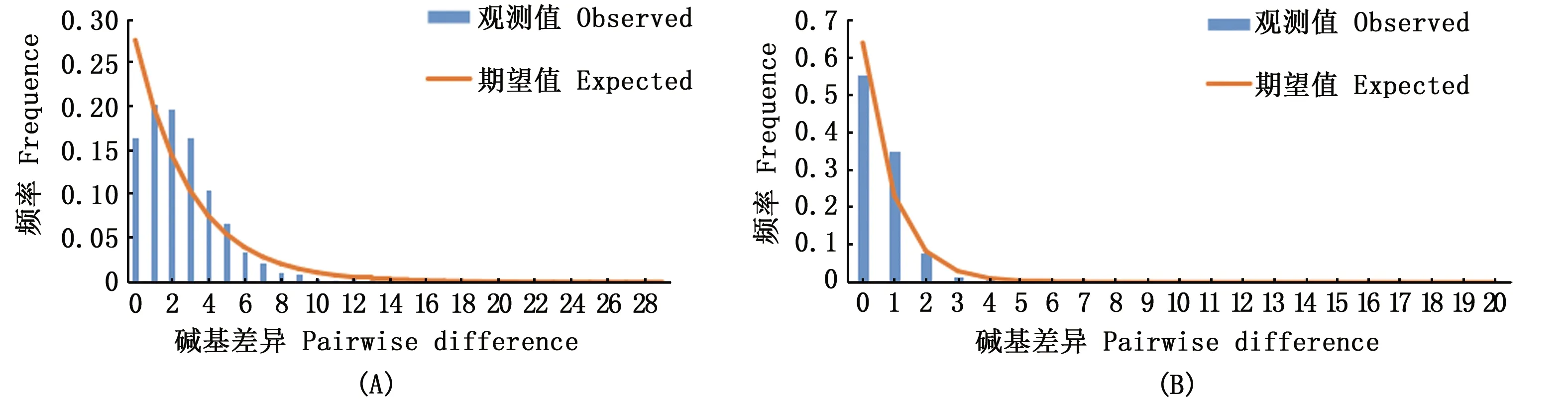
图3 基于Cytb (A)和D-Loop (B)序列的大獭蛤5个群体核苷酸不配对分布图Fig.3 Mismatch distribution of five populations based on Cytb (A) and D-Loop (B) sequences
3 讨论
遗传多样性又称基因多样性,生物种群的遗传多样性是其物种进化和环境适应的基础,遗传多样性缺乏可能会导致物种的资源衰退甚至濒临灭绝,丰富的遗传多样性能够确保物种的延续,同时也为物种进化提供充足的潜力。
3.1 遗传多样性
大獭蛤5个群体的线粒体Cytb基因序列中,检测到89个多态位点和73个单倍型,单倍型多样性为0.834 5±0.030 3;D-Loop基因序列中,检测到25个多态位点和27个单倍型,单倍型多样性为0.445 6±0.049 8,整体呈现出中等偏高的遗传多样性,但Cytb和D-Loop的核苷酸多样性仅分别为0.002 3±0.001 4和0.001 4±0.001 3,处于一个较低水平。一般来说,当群体数量足够大并且在很长一段时间内保持稳定,该群体中的高水平遗传变异是可以维持的,而当群体经历严重的瓶颈效应时,随机遗传漂变将导致遗传变异的大量丧失[11]。根据GRANT和BOWEN[32]提出的分类标准,以Hd=0.5、Pi=0.005为界,本研究结果与高单倍型多样性和低核苷酸多样性的模式相符。该遗传多样性模式的种群在历史上有经历扩张的可能,即某种群在数量急剧减少后,存留的优良个体历经瓶颈效应或建群效应,突然快速繁衍发展为一个大种群的现象,在扩张过程中,种群数量的增加导致了单倍型多样性的提高,而无充足的时间去积累核苷酸变异,所以会产生单倍型多样性较高而核苷酸多样性较低的遗传多样性模式[33]。
基于Cytb基因获得的5个大獭蛤地理群体的总体遗传多样性指数大于D-Loop序列的结果,通常情况下D-Loop序列获得的种群遗传变异大于Cytb基因[10]。目前有人发现mtDNA控制区D-Loop变异速率低于Cytb的现象,赵亮等[34]对太湖新银鱼(Neosalanxtaihuensis)Cytb和D-Loop序列采用贝叶斯(Bayes)MCMC模拟估测出Cytb相对速率为1.000±0.131,而D-Loop相对速率为0.859±0.261,Cytb的进化速率相对比D-Loop高。
3.2 种群遗传结构
遗传距离是反映不同物种或不同种群之间变异的常用参数。5个群体之间的遗传距离几乎处于同一水平,说明各群体之间的基因交流不受影响。ARNAUD等[35]认为在幼虫期具有浮游特性的海洋双壳类生物具有很强的分散能力,大獭蛤具有大约12 d的浮游期[18],在此期间一定程度上受到我国东南海暖流和东南部沿岸流的影响[36],从而被动地随着海流扩散和基因交流;此外我国大獭蛤苗种培育[19]的成功和工厂化育苗技术[7]的日渐成熟,导致各地大獭蛤群体之间交流加剧。
遗传分化指数Fst是作为对种群遗传分化评估的另一个重要指标。基于Cytb的Fst值在-0.008 0~0.010 1之间,基于D-Loop的Fst值在-0.014 3 ~ -0.007 2之间,Fst值结果也显示5个群体间存在负值现象,表明群体内差异大于群体间差异,同时根据WRIGHT[29]定义的标准和AMOVA分析结果表明,群体内的变异远大于群体间的变异,说明各地理群体间不存在显著的遗传分化。基于Cytb基因和D-Loop序列构建的NJ树中,5个群体的单倍型相互混杂,未形成特定的地理聚群。这些结果表明各区域个体间的交配是随机的,没有显著的遗传分化。黄荣莲等[37]在对贻贝(Pernaviridis)的研究中发现环境胁迫会对群体结构产生影响。本研究采样点福州(26°08′ N、119°30′ E)、厦门(24°48′ N、118°09′ E)和北海(21°48′ N、109°12′ E)为亚热带季风性气候;湛江(21°27′ N、110°37′ E)和涠洲岛(21°04′ N、109°12′ E)为热带季风性气候,5个采样地点的气候、温度和盐度等环境因素都大致相同,这可能是遗传分化弱的原因之一。
3.3 种群历史动态
通过分析种群的核苷酸不配对分布曲线的单峰或多峰类型,以及是否符合中性检验,来推断大獭蛤种群近期是否出现过种群扩张事件[38]。当种群偏离中性检验,Tajima’D检验会给出一个较低值。Fu’sFS值的负数越大表明由于种群扩张或种群选择导致中性偏离。整体来说,基于大獭蛤Cytb和D-Loop的Tajima’D和Fu’sFS值均为负值,且统计检验显著(P<0.05),核苷酸错配分布曲线为单峰,说明大獭蛤种群历史上可能经历过瓶颈和种群扩张,上述的高单倍型多样性和低核苷酸多样性的分布模式也证明了这一点。根据平均τ值估算的大獭蛤种群扩张时间距今约为1.3×104~ 6.6×104年,这个时期属于更新世晚期,中国沿海海洋鱼类的种群扩张大多发生在该时期[39]。该时期由于冰期和间冰期的交替,导致海平面剧烈变化,使得海平面下降120 ~ 140 m,现生海洋物种在近期历史上的扩张大多受到这种冰期和间冰期交替的影响[40-41]。因此,大獭蛤种群可能经历了同样的种群扩张。
综上所述,本研究基于线粒体Cytb和D-Loop序列对大獭蛤5个地理群体的遗传多样性进行了分析。整体来说,各群体间遗传距离较小,遗传分化程度较弱,遗传变异水平较低,可作为一个管理保护单位,且近期发生过种群扩张事件;结合群体内遗传距离和平均遗传多样性指数,可以得出大獭蛤的遗传多样性处于一个中等的水平。但近年来东南沿海地区生态环境破坏的加剧和人工繁殖技术的日臻成熟,势必会对大獭蛤的种质资源造成潜在的威胁。因此在合理的开发利用下,有必要对野生资源进行保护,以保证其行业的可持续性发展。
