一测多评法同时测定小儿金翘颗粒中7种成分的含量
2020-06-09梁欣雨周晓钢何兵杨世艳
梁欣雨 周晓钢 何兵 杨世艳
中圖分类号 R917 文献标志码 A 文章编号 1001-0408(2020)10-1179-06
DOI 10.6039/j.issn.1001-0408.2020.10.05
摘 要 目的:建立同时测定小儿金翘颗粒中7种成分含量的方法。方法:采用一测多评法(QAMS)。色谱柱为Lubex Kromasil C18,流动相为乙腈-0.1%磷酸水溶液(梯度洗脱),检测波长为326 nm,流速为1.0 mL/min,柱温为40 ℃,进样量为10 ?L。以绿原酸为内参物,计算新绿原酸、隐绿原酸、连翘酯苷A、异绿原酸B、异绿原酸A、异绿原酸C的相对校正因子;考察不同色谱系统、色谱柱、流动相比例、流速、柱温等对相对校正因子的影响,并采用两点校正法结合相对保留时间校正法对各成分进行色谱峰定位。分别按QAMS法和标准曲线法检测7种成分的含量,并进行比较。结果:新绿原酸、绿原酸、隐绿原酸、连翘酯苷A、异绿原酸B、异绿原酸A、异绿原酸C检测质量浓度的线性范围分别为9.27~92.70、37.36~373.60、13.02~130.20、7.15~71.50、4.56~45.60、6.32~63.20、14.69~146.90 ?g/mL(r≥0.999 7);定量限分别为1.38、1.41、1.40、1.99、1.10、1.17、1.10 ng,检测限分别为0.41、0.42、0.42、0.60、0.33、0.35、0.33 ng;精密度、重复性、稳定性试验的RSD均小于2%,平均加样回收率为98.28%~99.15%(RSD<2%,n=9)。新绿原酸、隐绿原酸、连翘酯苷A、异绿原酸B、异绿原酸A、异绿原酸C相对于绿原酸的相对校正因子分别为0.995、1.007、0.580、1.243、1.252、1.247;不同色谱条件下其相对校正因子的RSD均小于3%。采用两点校正法结合相对保留时间校正法预测的各待测成分的保留时间与实测值的相对误差绝对值均小于2%。QAMS法测得的含量分别为2.790~3.416、14.526~17.907、3.763~4.531、1.625~1.982、1.087~1.523、1.434~2.219、3.631~5.078 mg/g,标准曲线法测得的含量分别为2.811~3.438、14.512~17.893、3.739~4.508、1.656~2.012、1.108~1.544、1.460~2.245、3.597~5.045 mg/g;两种方法测得含量的相对误差均在±2%以内。结论:两点校正法结合相对保留时间校正法能准确定位各成分色谱峰;所建QAMS简便、快捷、准确、可靠,可用于同时测定小儿金翘颗粒中7种成分的含量。
关键词 一测多评法;小儿金翘颗粒;含量测定;色谱峰定位;两点校正法;相对保留时间校正法
Simultaneous Determination of 7 Constituents in Xiaoer Jinqiao Granules by QAMS
LIANG Xinyu1,ZHOU Xiaogang2,HE Bing2,YANG Shiyan3(1. The First Clinical College of Chongqing Medical University, Chongqing 400016, China; 2. School of Pharmaceutical Sciences, Southwest Medical University, Sichuan Luzhou 646000, China; 3. Dept. of Ultrasonic Imaging, the Affiliated Traditional Chinese Medicine Hospital of Southwest Medical University, Sichuan Luzhou 646000, China)
ABSTRACT OBJECTIVE: To establish a method for simultaneous determination of 7 constituents in Xiaoer jinqiao granules. METHODS: QAMS method was adopted. The determination was performed on Lubex Kromasil C18 column with mobile phase consisted of acetonitrile-0.1% phosphoric acid solution (gradient elution). The detection wavelength was set at 326 nm, the flow rate was 1.0 mL/min. The column temperature was 40 ℃, and sample size was 10 ?L. Using chlorogenic acid as the internal reference, the relative correction factors (RCF) of neochlorogenic acid, cryptochlorogenic acid, forsythiaside A, isochlorogenic acid B, isochlorogenic acid A and isochlorogenic acid C were calculated. The effects of different chromatogram system, chromatogram column, the ratio of mobile phase, flow rate and column temperature on RCF were investigaten. According to the two-point correction method combined with the relative retention time correction of the components to be tested, the peak location was carried out. The contents of 7 components were determined by QAMS and SCM respectively and then compared. RESULTS: The linear range of neochlorogenic acid, chlorogenic acid, cryptochlorogenic acid, forsythoside A, isochlorogenic acid B, isochlorogenic acid A, isochlorogenic acid C were 9.27-92.70, 37.36-373.60, 13.02-130.20, 7.15-71.50, 4.56-45.60, 6.32-63.20, 14.69-146.90 ?g/mL (r≥0.999 7). The limits of quantification were 1.38, 1.41, 1.40, 1.99, 1.10, 1.17, 1.10 ng, respectively; and the limits of detection were 0.41, 0.42, 0.42, 0.60, 0.33, 0.35, 0.33 ng, respectively. RSDs of precision, repeatability and stability tests were all less than 2%; average recoveries were 98.28%-99.15% (RSD<2.0%, n=9). RCFs of neochlorogenic acid, cryptochlorogenic acid, forsythiaside A, isochlorogenic acid B, isochlorogenic acid A and isochlorogenic acid C relative to chlorogenic acid were 0.995, 1.007, 0.580, 1.243, 1.252 and 1.247, respectively. RSDs of RCFs were all lower than 3% under different chromatogram conditions. Absolute value of relative error between the relative retention time of components to be tested predicted by two-point correction combined with relative retention time correction and measured value was less than 2%. The contents measured by QAMS were 2.790-3.416, 14.526-17.907, 3.763-4.531, 1.625-1.982, 1.087-1.523, 1.434-2.219, 3.631-5.078 mg/g; the contents measured by SCM method were 2.811-3.438, 14.512-17.893, 3.739-4.508, 1.656-2.012, 1.108-1.544, 1.460-2.245, 3.597-5.045 mg/g; relative errors of the two methods were within±2%. CONCLUSIONS: Two-point correction method combined with relative retention time correction can accurately locate the peaks of each constituent; established QAMS method is simple, rapid, accurate and reliable, it can be used for simultaneous determination of 7 constituents in Xiaoer jinqiao granules.
KEYWORDS QAMS; Xiaoer jinqiao granules; Content determination; Peak location; Two-point correction method; Relative retention time correction
小儿金翘颗粒是由金银花、连翘、葛根、大青叶、山豆根、柴胡、甘草等中药组成的复方制剂,具有疏风清热、解毒利咽、消肿止痛之功效[1],临床上主要用于治疗小儿急性上呼吸道感染、流行性感冒、急性扁桃体炎、小儿手足口病等[2-5]。该制剂中金银花为君药,含有绿原酸、新绿原酸、隐绿原酸和异绿原酸A、B、C等多种酚酸类成分,具有较强的抗菌、抗炎、抗氧化等生物活性[6]。连翘酯苷A是臣药连翘的主要活性成分,具有抗菌、抗病毒、抗氧化等药理活性[7]。《国家药品标准:新药转正标准(第66册)》[1]中记载,小儿金翘颗粒以绿原酸为质量控制指标,采用高效液相色谱法(HPLC)检测。虽然也有文献报道了小儿金翘颗粒的质量控制,但均以绿原酸或连翘苷含量为指标[8-9]。由于绿原酸类成分性质不稳定,在储存过程中可与其异构体发生相互转化[10],因此以绿原酸单一成分作为评价指标具有一定的片面性,无法准确反映制剂的质量。连翘的有效成分连翘苷含量较低,且在本研究前期试验中发现,其在小儿金翘颗粒中难以检测,故连翘苷不宜作为评价指标,而宜选择含量更高且更稳定的活性成分连翘酯苷A作为指标成分。
在中药多指标定量分析中,由于部分对照品紧缺、昂贵,导致传统多指标定量检测成本较高。而一测多评法(QAMS)能以一种便宜、易得的对照品为内参物,通过相对校正因子测定其他成分的含量[11]。QAMS法的关键是如何准确定位各成分的色谱峰,但由于仪器、色谱柱不同,待测成分的保留时间会发生明显飘移,从而影响其准确定位。目前,常用的色谱峰定位方法包括相对保留时间校正法和两点校正法,其中相对保留时间校正法仅适用于保留时间靠前的色谱峰,随着保留时间的增加,其定位误差越大[11];两点校正法适用于相对保留时间靠后的色谱峰,但唯一不足的是保留时间靠前的(特别是第1个)色谱峰定位不准[11]。基于此,本研究采用QAMS法同时测定小儿金翘颗粒中绿原酸、新绿原酸、隐绿原酸、异绿原酸A、异绿原酸B、异绿原酸C、连翘酯苷A的含量;通过两点校正法结合相对保留时间校正法对待测成分进行色谱峰定位,旨在为进一步完善和提高该药的质量标准提供参考。
1材料
1.1 仪器
P680-PDA型HPLC仪,包括四元梯度泵、二极管阵列检测器、柱温箱和色谱工作站(美国Dionex公司);LC2030型HPLC仪,包括四元梯度泵、紫外检测器、自动进样器、柱温箱和色谱工作站(日本Shimadzu公司);MS105DU型十万分之一电子天平(瑞士Mettler-Toledo公司);AS7240A型超声波清洗器(天津奥特赛恩斯仪器有限公司);Direct-Q3型超纯水系统(美国Millipore公司)。
1.2 药品与试剂
小儿金翘颗粒(四川凯京制药有限公司,批号:20180912、20181106、20181228,规格:5 g/袋);小儿金翘阴性对照颗粒(由本课题组按该制剂的处方工艺分别制得的缺金银花、连翘的阴性样品);新绿原酸对照品(批号:A0023-19031001,纯度:99.67%)、绿原酸对照品(批号:A0022-19030620,纯度:99.39%)、隐绿原酸对照品(批号:A0024-19032403,纯度:99.07%)、异绿原酸B对照品(批号:A0026-19031602,纯度:99.05%)、异绿原酸A对照品(批号:A0025-19032601,纯度:98.82%)、异绿原酸C对照品(批号:A0027-19031603,纯度:99.84%)、连翘酯苷A对照品(批号:A0590-18061301,纯度:99.15%)均购自成都曼斯特生物科技有限公司;乙腈、甲醇为色谱纯,其余试剂均为分析纯,水为超纯水。
2 方法与结果
2.1 色谱条件
色谱柱:Lubex Kromasil C18(250 mm×4.6 mm,5 ?m);流动相:乙腈(A)-0.1%磷酸水溶液(B),梯度洗脱(0~5 min,10%A→15%A;5~15 min,15%A→30%A;15~16 min,30%A→10%A;16~20 min,10%A);检测波长:326 nm;流速:1.0 mL/min;柱温:40 ℃;进样量:10 ?L。
2.2 溶液的制备
2.2.1 混合对照品溶液 分别精密称取新绿原酸、绿原酸、隐绿原酸、连翘酯苷A、异绿原酸B、异绿原酸A、异绿原酸C对照品适量,加75%甲醇溶解,制成上述7种成分质量浓度分别为0.927、3.736、1.302、0.715、0.456、0.632、1.469 mg/mL的单一对照品贮备液。取上述各单一对照品贮备液0.8 mL,置于10 mL量瓶中,加75%甲醇稀釋至刻度,摇匀,制成上述7种成分质量浓度分别为的0.074、0.299、0.104、0.057、0.036、0.051、0.118 mg/mL混合对照品溶液,避光保存。
2.2.2 供试品溶液 取本品适量,研细,取约1.0 g,精密称定,置于具塞锥形瓶中,精密加入75%甲醇50 mL,称定质量,超声(功率:240 W,频率:40 kHz)处理10 min,冷却至室温,再次称定质量,用75%甲醇补足减失的质量,摇匀,滤过,取续滤液,即得。
2.2.3 阴性对照溶液 取小儿金翘阴性对照颗粒,按“2.2.2”项下方法分别制备缺金银花、连翘的阴性对照溶液。
2.3 系统适用性试验
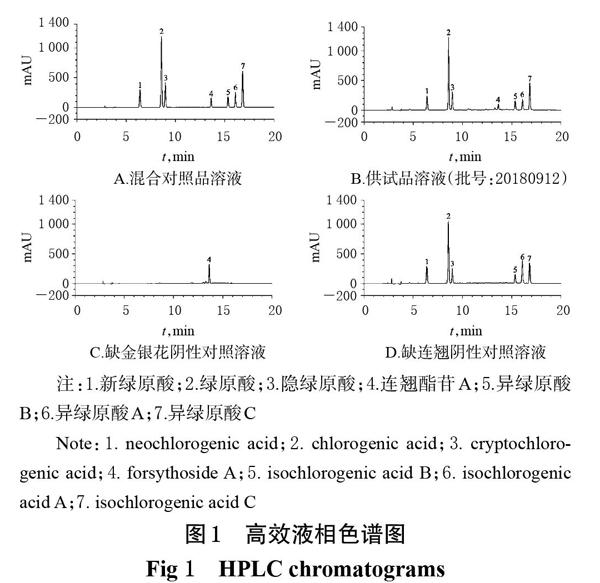
分别精密量取“2.2”项下混合对照品溶液、供试品溶液、阴性对照溶液适量,按“2.1”项下色谱条件进样测定,记录色谱图,详见图1。结果,新绿原酸、绿原酸、隐绿原酸、连翘酯苷A、异绿原酸B、异绿原酸A、异绿原酸C与其相邻色谱峰的分离度均大于2.22;拖尾因子分别为1.07、1.00、1.03、1.01、1.00、1.07、1.10,理论板数均不低于23 000;阴性对照在对应位置处无吸收峰,对测定无干扰。
2.4线性关系与定量限、检测限考察
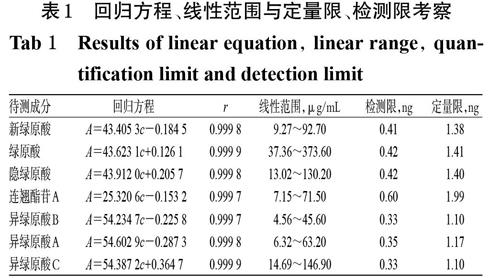
分别精密量取“2.2.1”项下各单一对照品贮备液0.1、0.2、0.4、0.6、0.8、1 mL,置于10 mL量瓶中,加75%甲醇稀释至刻度,制得系列线性工作溶液,按“2.1”项下色谱条件进样测定,记录峰面积。以待测成分质量浓度(c,?g/mL)为横坐标、峰面积(A)为纵坐标进行线性回归,回归方程与线性范围见表1。另取“2.2.1” 项下混合对照品溶液适量,用75%甲醇逐级稀释,分别以信噪比10 ∶ 1、3 ∶ 1计算定量限、检测限,结果见表1。
2.5 精密度试验
取“2.2.1”项下混合对照品溶液适量,按“2.1”项下色谱条件连续进样测定6次,记录峰面积。结果,新绿原酸、绿原酸、隐绿原酸、连翘酯苷A、异绿原酸B、异绿原酸A、异绿原酸C峰面积的RSD分别为0.29%、0.17%、0.32%、0.41%、0.36%、0.33%、0.25%(n=6),表明仪器精密度良好。
2.6 重复性试验
取小儿金翘颗粒样品(批号:20180912),每份1.0 g,共6份,精密称定,按“2.2.2”项下方法制备供试品溶液,再按“2.1”项下色谱条件进样测定,记录峰面积并按标准曲线法计算样品中7种待测成分的含量。结果,新绿原酸、绿原酸、隐绿原酸、连翘酯苷A、异绿原酸B、异绿原酸A、异绿原酸C含量的RSD分别为1.19%、1.08%、1.26%、1.41%、1.32%、1.20%、1.13%(n=6),表明本方法重复性良好。
2.7 稳定性试验
取 “2.2.2”项下供试品溶液(批号:20180912)适量,在室温密闭条件下放置0、4、8、12、24、36 h时,按“2.1”项下色谱条件进样测定,记录峰面积。结果,新绿原酸、绿原酸、隐绿原酸、连翘酯苷A、异绿原酸B、异绿原酸A、异绿原酸C峰面积的RSD分别为0.83%、0.66%、0.95%、0.90%、0.87%、0.78%、0.71%(n=6),表明供试品溶液在室温密闭条件下放置36 h内稳定性良好。
2.8 加样回收率试验
取已知含量的小儿金翘颗粒(批号:20180912),每份0.5 g,共9份,精密称定,分别加入一定量的单一对照品溶液(按“2.2.1”项下方法制备,含新绿原酸1.925 mg/mL、绿原酸10.219 mg/mL、隐绿原酸2.507 mg/mL、连翘酯苷A 1.094 mg/mL、异绿原酸B 1.016 mg/mL、异绿原酸A 1.312 mg/mL、异绿原酸C 2.968 mg/mL),按“2.2.2”项下方法制备供试品溶液,再按“2.1”项下色谱条件进样测定,记录峰面积并计算加样回收率。结果,新绿原酸、绿原酸、隐绿原酸、连翘酯苷A、异绿原酸B、异绿原酸A、异绿原酸C的平均加样回收率分别为98.63%(RSD=1.39%,n=9)、99.15%(RSD=1.10%,n=9)、98.78%(RSD=1.34%,n=9)、98.37%(RSD=1.53%,n=9)、98.28%(RSD=1.62%,n=9)、98.46%(RSD=1.50%,n=9)、98.85%(RSD=1.19%,n=9)。
2.9 相对校正因子的计算及验证
2.9.1 相对校正因子的计算 本研究前期试验发现,由于小儿金翘颗粒中绿原酸含量最高,其对照品价格便宜且易得,故以绿原酸为内参物,计算新绿原酸、隐绿原酸、连翘酯苷A、异绿原酸B、异绿原酸A及异绿原酸C相对于绿原酸的相对校正因子(fi/s),相对校正因子(fi/s)=ai/as[12]。式中,ai为待测成分标准曲线斜率,as为内参物标准曲线低利率。结果,新绿原酸、隐绿原酸、连翘酯苷A、异绿原酸B、异绿原酸A及异绿原酸C相对于绿原酸的相对校正因子分别为0.995、1.007、0.580、1.243、1.252、1.247。
2.9.2 相对校正因子耐用性考察 为考察相对校正因子在不同色谱条件下的耐用性,本研究适当改变了流动相各梯度点的比例(±2%)、流速(±2%)、柱温(±5 ℃)、水相pH(±0.2)、检测波长(±5 nm),并平行操作3次。结果,各待测成分相对校正因子的RSD均小于3%(n=3),表明色谱条件在一定范围内的变化对各待测成分相对校正因子的影响较小。
2.9.3 相对校正因子重现性考察 本研究考察了不同品牌HPLC仪(Dionex、Shimadzu)以及不同色谱柱[Lubex Kromasil C18、AkzoNobel Kromasil C18、Dikma Silversil C18,均为(250 mm×4.6 mm,5 ?m)]对相对校正因子的影响,平行操作3次。结果,各待测成分相对校正因子的RSD均小于2%(n=3),表明相對校正因子在不同色谱条件下具有良好的重现性。
2.10 待测成分色谱峰的定位
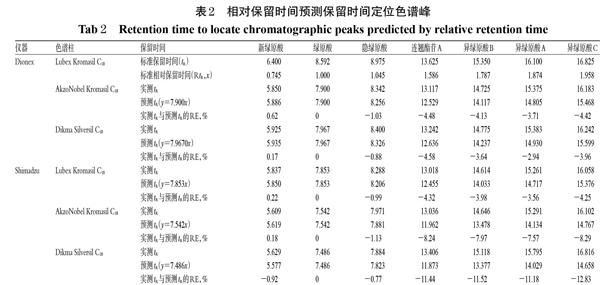
QAMS法通常采用相对保留时间定位色谱峰,即通过对比不同条件下的相对保留时间,若相对保留时间基本一致则认定为同一色谱峰[13]。当色谱峰众多且密集时,由于未测量的其余色谱峰均需计算相对保留时间,因此其操作非常繁琐。假设其余条件下的相对保留时间与标准相对保留时间一致,再计算其相对应的理论保留时间,则可简化过程,即各色谱峰的预测保留时间(y)=其余条件下内参物绿原酸的保留时间×Lubex Kromasil C18对应的标准相对保留时间(x),结果见表2[以第1根色谱柱为标准,相对误差(RE)=(预测保留时间-实测保留时间)/实测保留时间×100%]。
由表2可知,采用相对保留时间校正法定位色谱峰时,保留时间越靠前的色谱峰,其实测和预测保留时间的RE绝对值越小,定位越准确,这部分色谱峰可采用相对保留时间校正法定位;但随着保留时间的增加,保留时间靠后的色谱峰的实测和预测保留时间的RE绝对值越大,保留时间甚至相差2 min以上,难以准确定位。
有研究发现,在相同的色谱条件下即使采用不同的仪器和色谱柱,各成分的保留时间也存在简单的线性关系[14]。根据此原理,本研究以待测成分在Lubex Kromasil C18的保留时间为横坐标(x),以其余色谱柱的保留时间为纵坐标(y),绘制标准曲线。结果发现,Lubex Kromasil C18与其余色谱柱保留时间的相关系数分别为0.999 7、0.999 4、0.999 7、0.999 0、0.997 8。由于本研究的目的仅为定位色谱峰,且在实际操作中,因其他色谱峰未知,故无法采用多个数据点计算保留时间的标准曲线,但若在色谱图中前后各取一点,以内参物绿原酸和最易辨认的色谱峰(如最后一个色谱峰异绿原酸C)进行两点校正[15],同法绘制校正曲线。结果发现,两点校正与7个点绘制的标准曲线基本重合,提示两点校正法可用于预测各成分的出峰时间,随后将Lubex Kromasil C18的标准保留时间带入校正方程,计算各成分的预测保留时间,结果见表3。
由表3可知,即使采用不同的仪器和色谱柱,随着保留时间的增加,采用两点校正法的预测保留时间和实测保留时间的RE绝对值较低,且非常接近。但若选择的点不包含第1个色谱峰,则第1个色谱峰的定位不准。因此对于第1个色谱峰新绿原酸,可采用相对保留时间校正法预测定位,其余成分的色谱峰可采用两点校正法预测定位,这可使每个色谱峰均能获得准确的预测保留时间(RE<2%)。若色谱峰临近处还有其他色谱峰,则可根据各成分的紫外吸收光谱(图2)、整体峰形或峰面积百分比进一步确认[15]。
2.11 含量测定结果比较
分别采用QAMS法和标准曲线法测定样品中各成分的含量,平行操作3次,结果见表4。结果显示,2种方法测得含量的RE绝对值均小于2%,表明两者测定结果基本一致,QAMS法可用于小儿金翘颗粒中多指标成分的测定[RE=(QAMS法测得值-标准曲线法测得值)/标准曲线法测得值×100%]。
3 讨论
3.1 相对校正因子计算
本研究采用斜率校正法计算相对校正因子,即以各待测成分与内参物标准曲线斜率之比作为校正因子,该方法相对于多点校正更加简便、快捷[12]。采用斜率校正法的结果与标准曲线法结果之间的差异主要由斜率截距造成,故当标准曲线线性关系较好,斜率截距之比>100时,二者之间的差异才能足够小,以斜率校正法计算校正因子才具有较好的可行性和准确性[16]。
3.2 色谱峰的定位
有研究认为,在色谱峰定位时,以各条件下相对保留时间的RSD<5%作为色谱峰准确定位的依据[11]。但本研究結果发现,即使满足该条件,保留时间靠后的色谱峰,其预测保留时间和实测保留时间仍相差较大(RE绝对值>10%),且部分色谱峰实测与预测保留时间相差超过2 min。这提示在色谱峰较多、密集且色谱峰紫外吸收相似时,特别是在批间整体峰形不规则的中草药中采用相对保留时间校正法很难准确定位各色谱峰。本研究采用两点校正获得校正方程,通过方程预测各条件下的理论出峰时间,从而快速定位色谱峰。结果发现,除第1个色谱峰外,其余色谱峰均可准确定位。因此,本研究以相对保留时间校正法定位新绿原酸峰,以两点校正法定位其他色谱峰。结果显示,所有色谱峰预测和实测保留时间的RE绝对值均小于2%,预测与实测保留时间相差小于0.3 min,提示所有色谱峰均可实现准确定位。
3.3 准确性评价
目前,评价QAMS法准确性的方法较多,如t检验、方差分析、相关系数、夹角余弦等[17]。为验证上述方法的可靠性,本研究前期模拟了因校正因子不准确所致的较大测量误差,即将QAMS法测得的任意一个测定值或者所有值同时缩放10倍后再与标准曲线法进行t检验和方差分析。结果发现,其P值仍大于0.05,无显著性差异;随后,将任意一个测定值缩放10%或所有测定值同时缩放10倍后发现,相关系数或夹角余弦依然>0.999,仍然具有极高的相似性。因此,本研究在参考相关文献[18],采用RE评价QAMS法的准确性,即任一测定值超过标准曲线法测定值的±2%时,其RE绝对值>2%,提示采用相对误差评价其准确性更加科学合理。
3.4 含量测定结果分析
本研究结果显示,2种方法测得含量的RE绝对值均小于2%,提示2种方法的测定结果基本一致。QAMS法可用于同时测定小儿金翘颗粒中7种成分的含量。
4 结语
本研究所建两点校正法结合相对保留时间校正法能准确定位各成分色谱峰;QAMS法简便、快捷且准确、可靠,可用于同时测定小儿金翘颗粒中7种成分的含量。
参考文献
[ 1 ] 国家药典委员会.国家药品标准:新药转正标准:第66册[S].北京:化学工业出版社,2008:46-49.
[ 2 ] 尹经典.小儿金翘颗粒治疗小儿急性上呼吸道感染的临床疗效观察[J].世界最新医学信息文摘,2019,19(50):174、178.
[ 3 ] 黄鹤.小儿金翘颗粒治疗小儿流行性感冒的临床观察[J].世界最新医学信息文摘,2019,19(50):177-178.
[ 4 ] 吴香君,王丹英.小儿金翘颗粒辅助治疗小儿急性化脓性扁桃体炎49例临床观察[J].中医儿科杂志,2017,13(3):34-37.
[ 5 ] 贾东新,曹丽娜,王文霞.小儿金翘颗粒治疗小儿手足口病的疗效分析[J].山西医药杂志,2013,42(1):73-74.
[ 6 ] 那袭雪,张文涛,谈远锋,等.绿原酸及其异构体药理作用及不良反应研究进展[J].辽宁中医药大学学报,2018,20(3):140-144.
[ 7 ] 付鹏亮,王东强,李志军.连翘酯苷药理作用研究进展[J].长春中医药大学学报,2011,27(6):1062-1063.
[ 8 ] 杨丽波,黄丽,杜宏,等.反相高效液相色谱法测定小儿金翘颗粒中绿原酸的含量[J].大理学院学报,2012,11(9):98-100.
[ 9 ] 李志成,邓涛,杨永寿. HPLC法测定小儿金翘颗粒中连翘苷的含量[J].大理学院学报,2012,11(6):23-25.
[10] 朱鹏,苗潇磊,陈勇.绿原酸、隐绿原酸和新绿原酸在中性和碱性pH条件下的降解动力学[J].药学学报,2016,51(1):122-126.
[11] 王智民,钱忠直,张启伟,等.一测多评法建立的技术指南[J].中国中药杂志,2011,36(6):657-658.
[12] 何兵,杨世艳,张燕,等.一测多评中待测成分校正和定位的新方法研究[J].药学学报,2012,47(12):1653-1659.
[13] 左雅敏,李琛,彭兴春,等. HPLC-一测多评法测定黄精及其饮片中6种成分的含量[J].中国药房,2019,30(13):1748-1754.
[14] 王龙星,肖红斌,梁鑫淼. 1种提高色谱指纹谱保留时间重现性的新方法[J].分析化学,2011,31(10):1232-1236.
[15] 何兵,刘艳,杨世艳,等. HPLC一測多评法同时测定双青咽喉片中10种成分[J].中草药,2013,44(8):974-981.
[16] 赵倩,冯伟红,张启伟,等.“一测多评”法用于栀子金花丸多成分含量测定的可行性研究[J].中国中药杂志,2014,39(10):1826-1833.
[17] 胡瑞雪,梁元昊,徐文丽,等.一测多评法在中药中的应用及研究进展[J].药物分析杂志,2019,39(11):1968-1979.
[18] 马昌豪,李怀伟,仝桂平,等.一测多评法同时测定咽炎片中5种指标性成分的含量[J].中国药房,2019,30(16):2232-2236.
(收稿日期:2020-01-09 修回日期:2020-04-07)
(编辑:陈 宏)
