高效液相色谱法测定阿苯达唑原料药的含量
2017-01-23张乐
张乐
摘 要: 为建立一种阿苯达唑原料药含量测定的高效液相方法,采用方法:色谱条件:Agilent ODS C18(5μm,3.0×150mm)色谱柱;以甲醇为流动相A;磷酸二氢钠水溶液(11g→800ml)为流动相B,甲醇—磷酸二氢钠水溶液=65:35时,流速为0.8ml/min,阿苯达唑对照品主峰保留时间为3.044min;分离度良好,扫描波长为292nm,柱温:30℃,结果阿苯达唑原料及溶剂峰均得到良好分离。阿苯达唑在2μg/ml~1200μg/ml浓度范围内线性良好(R2=0.99999,n=6),平均回收率98.0%,RSD为0.5%,定量限为0.1μg/ml,并得出结论:高效液相色谱法灵敏、准确,专属性强,重现性好,可用于阿苯达唑原料药含量的测定及质量控制。
关键词: 阿苯达唑 高效液相色谱法 紫外分光光度法 含量测定
阿苯达唑(Abendazole)是一种抗寄生虫的广谱药物,对阿苯达唑线虫、棘球蚴、绦虫、吸虫等具有较好的驱除作用[1-2],在兽医临床广泛应用。我们通常会采用高氯酸滴定法和紫外分光光度法来测定阿苯达唑原料粉的含量,这两种方法在《中华人民共和国兽药典》[3]中都有提到,但高氯酸滴定法和紫外分光光度法[4]受环境因素的影响较大,使得结果的专属性较差,准确度偏低。并且在兽药厂生产时可以看到,有些不良商家以次充好,仅仅用滴定法或紫外的方法都不能准确、有效地检测原料含量,所以本文利用高效液相色谱法[5-6],建立一种检测阿苯达唑原料药的方法。本文通过实际应用表明该方法可以准确快速地测定阿苯达唑原料药的含量,并且更有效地排除其他外界因素的影响,更好地通过含量测定控制阿苯达唑原料药的质量。本方法专属性强,重现性高,可以为阿苯达唑原料药含量测定提供新的参考。
1.材料
1.1试剂。阿苯达唑对照品(中国药品生物制品检定所);阿苯达唑原料(连云港市亚辉医药化工有限公司,批号:9011237);甲醇,色谱级;磷酸二氢钠;高氯酸,分析纯;水,自制超纯水。
1.2仪器。LC-20A岛津高效液相色谱仪;Agilent ODS C18(5μm,3.0×150mm)色谱柱;FA1004型电子天平;μV-5500PC型紫外可见光光度计;DHG-9030A电热鼓风干燥箱。

2.方法与结果
2.1色谱条件与系统适用性试验。Agilent ODS C18(5μm,3.0×150mm)色谱柱;以甲醇为流动相A;磷酸二氢钠水溶液(11g→800ml)为流动相B,通过筛选不同比例的流动相,得出当甲醇—磷酸二氢钠水溶液=65:35时,流速为0.8ml/min,阿苯达唑对照品主峰保留时间为3.044min;拖尾因子1.00;分离度为3.3,表明主峰和杂质峰分离度良好,因此选择其作为最佳的色谱条件;通过紫外分光光度计在200nm~400nm处扫描,得出在292nm处有最大紫外吸收,故确定扫描波长为292nm;柱温:30℃;进样量:10μl。理论板数阿苯达唑峰计算不低于2000。
2.2对照品溶液的制备。取阿苯达唑对照品在电子天平上精确称定10mg置200ml量瓶中,移取5ml冰乙酸使对照品溶解,再用流动相稀释至刻度线处,将定容后的对照品溶液超声50min,配得0.05mg/ml对照品溶液。
2.3原料药供试品溶液的制备。称取阿苯达唑供试品原料0.1g,用流动相A稀释制成每1ml中约含1mg的溶液,摇匀。将摇匀定容的溶液超声10min,精确移取,用流动相B稀释制成1ml中约含0.05mg/ml的溶液。
2.4重复性实验。取0.05mg/ml阿苯达唑对照品溶液10μl,注入液相色谱仪,检测其峰面积,重复上述实验6次,计算6次峰面积的RSD值为0.4%,表示其重复性良好。
2.5稳定性实验。取0.05mg/ml阿苯达唑对照品溶液放置2、4、6、8、12、24、36、48h,注入液相色谱仪进样,计算各时间的色谱峰面积的RSD值为0.5%,说明在常温放置条件48小时内,溶液稳定性良好。
2.6定量限。将2.2项下的对照品溶液用无水乙醇逐级稀释,信噪比10:1时,检出浓度为0.1μg/ml为定量限。

2.7线性及范围。将2.2项下配制的阿苯达唑对照品溶液浓度为1、2、3、4、5、6、10μg/mL分别进样20μL测定峰面积,以浓度为横坐标,峰面积为纵坐标,绘制标准曲线。以对照品峰面积对其浓度进行线性回归,回归方程:Y=381274.4237X+5349.3,R2=0.99999,表明2μg/ml-1200μg/ml浓度范围内峰面积与浓度呈良好的线性关系。
2.8加样回收率。分别精确量取原料品溶液配制的1mg/ml供试品溶液3ml置6个100ml容量瓶中,再精密量取对照品溶液配制的1mg/ml对照品溶液2ml和5ml各3份,分别加入该6个容量瓶中,用无水乙醇稀释至刻度。超声15min,用0.45μl微孔滤膜过滤,进样为10μl,测定各自的阿苯达唑含量,计算回收率、平均回收率及RSD值,测定结果见表一:
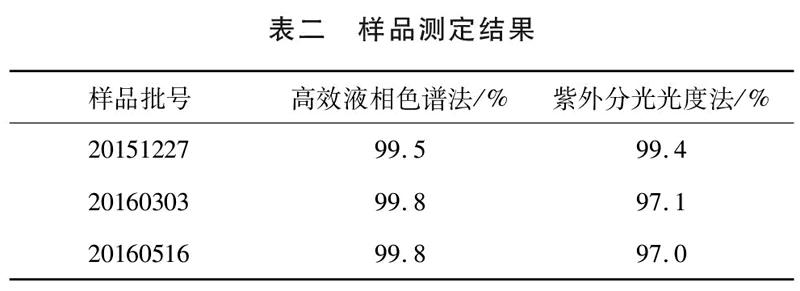
2.9含量测定。按2.3实验步骤配制0.10mg/ml供试品溶液,用外标法测定阿苯达唑含量(高效液相色谱图见图一,图二),并将测定结果紫外分光光度法测定结果进行对比。表二数据表明,我们所建立的高效液相色谱法能够准确测定阿苯达唑百分含量。
3.结果与讨论
3.1本文通过使用紫外分光光度法进行波长扫描,得出阿苯达唑原料在292nm处有最大吸收波长,从而作为最佳波长扫描范围。
3.2本文通过研究不同的流动相比例和流速,最终确定在甲醇—磷酸二氢钠水溶液=60:40时,流速为0.8ml/min,主峰和有关物质分离度良好,因此选择其作为最佳的色谱条件。
3.3本文所建立的方法优点在于:通过与标准品进行对照,专属性强,耗时短,出峰时间早,主峰和杂质峰分离度良好,可以很清晰地看出有关物质的存在,在兽药厂进行原料药检测时,可以很好地发现不良商家的以坏充好的意图。传统的滴定分析法和紫外分光光度法,仅说明被测物质可能是阿苯达唑,不能确定就是阿苯达唑原料,可能其他一类或混合物质也有可能具有相同的吸收波长,专属性和准确度都相对低于高效液相色谱法。本文建立的方法可以更准确地测定阿苯达唑原料药的含量及更好地控制阿苯达唑原料药的质量。
参考文献:
[1]Pourgholami MH,Woon SL,Almajd R,et al.In vitro and in vivo sup-pression of growth of hepatocellular carcinoma cells by albendazole[J].Cancer Lett,2001,165(1):43-49.
[2]Rolin S,Souhaili-el Amri H,Batt AM,et al.Study of the in vitro bioac-tivation of albendazole in human liver microsomes and hepatoma celllines[J].Cell biol Toxicol,1989,5(1):1-14.
[3]中国兽药典委员会.中华人民共和国兽药典(一部)[M].北京:中国农业出版社,2010:113-114.
[4]中国兽医药品监察所.兽药检验操作规程[M].北京:中国农业科技技术出版社,2005:66-68.
[5]中华人民共和国国家药典委员.中华人民共和国药典(二部)[S].北京:化学工业出版社,2015:557-558.
[6]刘志辉.HPLC法测定阿苯达唑的含量及有关物质[J].中国药师,2014(3):37-42.
