IDS 基因新发变异致黏多糖贮积症Ⅱ型1 例报告并文献复习
2020-06-06崔清洋李沙沙周福军曹银利
崔清洋 李沙沙 周福军 曹银利
新乡医学院第一附属医院儿科(河南卫辉 453100)
黏多糖贮积症(mucopolysaccharidosis,MPS)为一类罕见的溶酶体病,由于黏多糖降解酶的缺乏致酸性黏多糖不被全部降解,使黏多糖积聚于机体的不同组织,导致肝脾增大、智能障碍、骨骼畸形等一组临床症状及体征[1]。
MPS Ⅱ型又称作Hunter 综合征,于1917 年首次报道,美国报道发病率为1/25000 活产男婴,英国为l/132000 活产男婴[2],澳洲为l/165000 活产男婴[3],国内尚未见MPS Ⅱ型流行病学报道。MPS Ⅱ型在临床上属于罕见病,为亚洲最常见亚型,占MPS病例的50%,而在西方国家MPS Ⅰ型的发病率高于MPS Ⅱ型。MPS Ⅱ型发病的机制为IDS基因的缺陷,使艾杜糖醛酸硫酸酯酶减少,导致葡萄糖胺聚糖的降解障碍,使降解不完全的硫酸乙酰肝素和硫酸皮肤素大量的堆积在全身的各种脏器及组织中,出现相应的临床症状。
MPS Ⅱ型为一种严重的致残、致死性X连锁隐形遗传的单基因遗传病。罕见情况下,女性杂合的携带者也能出现临床症状。受累男性中,患者的发病年龄、症状严重程度和病程进展速度可有较大差异。早期发病者的神经系统受累往往表现为进行性认知功能下降。神经系统症状、呼吸系统通气障碍和心血管系统疾病可导致患者在10~20岁死亡[4]。慢性发病者的神经系统受累较轻,往往可以存活到成人期,且具有正常的认知功能[5]。本文回顾分析1例MPS Ⅱ型患儿的临床资料及基因检测的结果。
1 临床资料
患儿,男,5岁6月龄。因发现发育落后4年余入院,主要表现为言语较同龄儿童发育落后。G2P2,足月顺产,出生时无产伤及窒息史。父母体健,非近亲结婚。患儿哥哥10岁,外貌与患儿高度相似,罹患脑性瘫痪、脑积水,生活不能自理。体格检查:一般情况尚可,发育不良,营养中等,前额突出,眉毛浓密,眼睛突出;心肺查体未见异常;腹部膨隆,脐疝,肝肋下5 cm,质地中等,无压痛表现,脾脏肋下2 cm,无压痛表现;脊柱活动自如,双上肢活动受限,不能完全背伸,双下肢活动自如;四肢肌张力检查不合作,四肢肌力正常,生理反射存在,病理反射未引出。头颅磁共振成像示双侧额顶叶、半卵圆中心及侧脑室周围异常信号,扣带回多发性腔隙灶,脑沟、脑池及脑裂加深并脑室系统扩大。言语:可发出“走吧、买糖、找阿姨”;运动:不会单腿站,捏食指不能对捏、不会用筷子夹食物;认知:可认出“爸爸、妈妈、奶奶”等,会数数字到30,会写简单数字;社交:不容易与同龄儿童玩耍。孤独症行为评估量表得分54 分,高度怀疑孤独症谱系障碍。左手正位、左尺桡骨正位X线平片示左手第2~5掌骨近端变尖,第2~5近节及远节指骨末端稍变尖;左腕部见2枚腕骨影,左侧桡骨远端可见骨骺影,骨骺影较小,左侧尺骨远端未见骨骺影,左手第一掌骨近端可见骨骺影;对应骨龄为3.2岁男孩。见图1。腰椎生理曲度稍直,腰1 椎体稍前移,部分胸腰椎体上下缘膨隆,部分椎体前下角呈唇样改变,所示部分肋骨近端变细小,余所示骨质结构及椎间隙未见明显异常,考虑黏多糖症。结合患儿病史、体格检查及相关检查结果,考虑MPS可能性大。
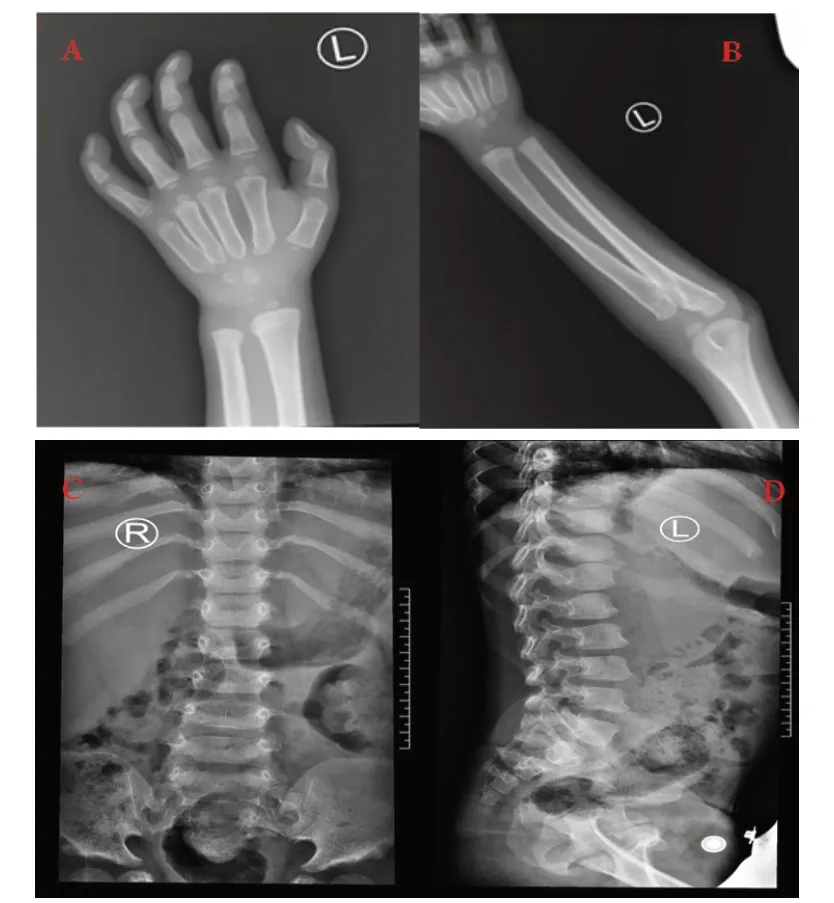
图2 患儿X 光片表现
经医学伦理审核及获得家长知情同意后,采集患儿外周血4 mL及其父母和哥哥外周血2 mL,乙二胺四乙酸(EDTA)抗凝,送北京的康旭医学检验有限公司进行基因检测。提取患儿及其父母和哥哥的基因组DNA,使用 Illumina 高通量测序仪和 Agilgent 公司的SureSelect 探针富集体系进行二代基因的检测。使用软件 CASAVA(1.8.2)将原始的数据转化为可识别的碱基序列,然后进行 Align分析、SNP 分析和 DIP 分析,获得靶向区域变异位点的信息。通过 PolyPhen-2、SIFT、Mutation Taster 进行蛋白质的损伤分析,获得需要进一步验证的变异位点。IDS 基因(exon 6)引物为:正向引物序列CCCCAGCACTTTGCCTGATA;反向引物序列GAGGTCCCTGATGGCCTACCPCR;扩增条件为:①预变性:95 ℃,10 min;②变性(35个循环):95 ℃,30 s;③退火(35个循环):60 ℃,30 s;④延伸(35个循环):72 ℃,45 s;⑤彻底延伸(35个循环):72 ℃,5 min。在人类的基因组数据库 GenBank 中获得IDS基因变异位点的基因序列,在引物设计的网站Primer Z(http://genepipe.ncgm.sinica.edu.tw/primerz/primerz 4.do)设计并合成引物。对变异的位点进行PCR 扩增后进行一代测序验证,排除掉二代测序中假阳性的位点。测序分析发现患儿IDS基因第6 号外显子上的c.820dupG(编码区第820号核苷酸G重复)的核苷酸变异,属于半合子变异,该变异导致从第274号氨基酸-谷氨酸开始的氨基酸合成发生改变,并在改变后的第 68个氨基酸终止(p.Glu274GlyfsTer68),为移码变异(见图2)。该变异可能导致蛋白质功能受到影响,该变异的致病性尚未见文献报道(所参考数据库:HGMD Pro及 PubMed)。对IDS基因进行蛋白质二级结构预测显示,包括10个α-螺旋,18个延伸链。各种二级结构所占的百分比为α-螺旋占16.36%,延伸链占8.18%,无规则卷曲占75.45%。c.820dupG(p.Glu274GlyfsTer68)变异使原来的酸性氨基酸谷氨酸变为中性甘氨酸氨基酸,推测该变异可能影响艾杜糖醛酸硫酸酯酶空间结构的稳定性。而该突变异为移码突变,导致从第274 号氨基酸谷氨酸开始的氨基酸合成发生改变,并在改变后的第68个氨基酸终止,推测会出现蛋白翻译错误而影响艾杜糖醛酸硫酸酯酶的功能。该变异不属于多态性变化,在人群中发生的频率极低(所参考数据库 1000 Genomes 及dbSNP)。家系验证结果显示,患儿半合子变异遗传自母亲,母亲为杂合变异,也即携带者,父亲未发现该基因位点的变异,也即野生型,患儿哥哥基因型同患儿,即男性半合子,符合X连锁隐性遗传规律。
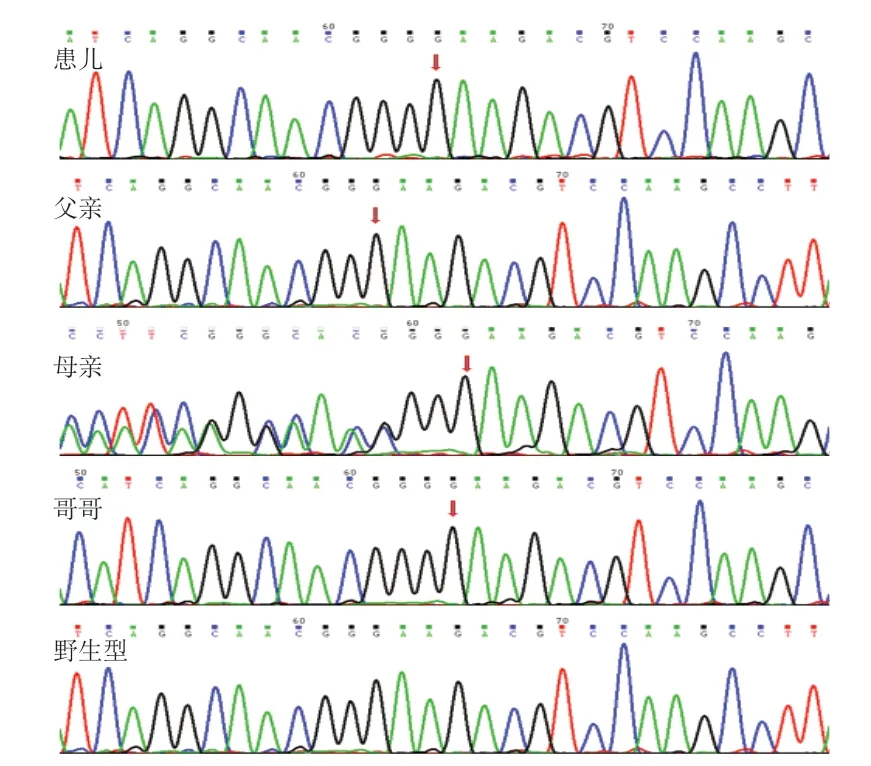
图2 IDS 基因c.820dupG 变异测序峰图
根据美国医学遗传学与基因组学学会(ACMG)联合美国分子病理学会(AMP)2015 年制订的“基因序列变异的解释标准和指南”进行致病性分析[6]。IDS基因 c.820dupG的致病性:①c.820dupG变异为移码变异,为致病变异(非常强致病性证据,PVS 1);②c.820dupG变异通过比照千人基因组数据库(1000 Genomes)、人类基因突变数据库(HGMD)未见其收录(中等致病性证据,PM2);③综合上述 c.820dupG变异的证据强度为“PVS 1+PM 2”,判断为可能是导致受检者发病的致病性变异。结合患儿临床表现及基因检测分析诊断IDS基因c.820dupG变异所致的MPSⅡ型基本明确,但患儿已经出院,告知患儿家属该病目前的酶替代疗法和骨髓移植方案,家属放弃。
2 讨论
编码艾杜糖醛酸硫酸酯酶的IDS基因定位于Xq27.32-Xq28,基因组全长约28.36 kb,包含9个外显子和8 个内含子,外显子长度约7619 bp,编码550个氨基酸[7]。截至 2018 年 3 月,人类基因突变数据库中的IDS基因突变的报道已经超过630 个。无义突变和错义突变是主要变异类型,而IDS基因点突变是非随机分布的,3、8、9号外显子变异最常见。此外,还包括剪接变异,小片段及大片段的插入、缺失等[8]。IDS基因有很高的突变率和高度遗传的异质性,与民族、种族的差异,IDS基因的结构及其所编码酶蛋白的结构的特殊性,基因所在X 染色体的结构的特殊性(含脆性部位)和X 染色体的随机的失活,突变热点、假的基因、隐蔽性的拼接位点、indel 的存在,和配子的形成过程中的复制、修复或交换过程中出现的偏差均可能有关[9]。在距离IDS基因的下游约20 kb的位置,有一个假的基因——IDS2,其内含子2、3、7的序列及外显子2、3 和IDS基因有明显的同源性,是为IDS基因造成突变和重组主要的原因[9]。大片段或整个IDS基因的缺失突变、IDS基因重排约占MPS Ⅱ型患者的19%~25%,小突变或微小病变 [包括插入、缺失、重复(<20 bp)等]约占MPS Ⅱ型的75%~80%[10]。
IDS基因的高度遗传异质性使得基因型和临床表型之间关系的建立较为困难,IDS基因的大片段缺失、基因-假基因重组的患者与重症临床表型相关,另外无义突变产生的终止密码子引起的表型通常被归为重型,国内报道的c.344 delA 的缺失变异和外显子4~6的大片段缺失变异者均是重型的MPS Ⅱ[11]。
IDS基因编码区上的所有变异位点不都具有致病性,迄今为止在编码区仅有2 个确认的多态性位点和1个疑似的多态性位点报道,分别是T146T、R313C、疑似的M 488 I,在表型健康的女性携带者中发现R 313 C,其母亲和姐姐均携带此多态性位点,该女性所生育的男孩也有该变异,但R313C在100个X染色体的对照样本中并未被发现,提示R313C是位于编码区的极其罕见的多态性位点[12-14]。
本例患儿的c.820dupG变异位点位于第6号外显子,与文献报道不同,c.820dupG(编码区第 820 号核苷酸 G 重复)的核苷酸变异导致从第 274 号谷氨酸开始的氨基酸合成发生改变,并在改变后的第68个氨基酸终止,为移码变异,转录提前终止,推测截短的氨基酸肽链影响了蛋白质的功能,后者缺乏硫酸酯酶的活性,进而使硫酸皮肤素和硫酸乙酰肝素不能有效的降解,在全身的脏器和组织内沉积导致严重的表型。
对欧洲30 个MPS Ⅱ家系的研究发现,母亲是家系中的第1 个带有变异基因的人,但通过单倍型的分析发现,患者母亲携带的变异基因的X染色体来自患者的外祖父,而其外祖父的相同X 染色体上的 IDS 基因无相应的变异,从而推测变异可能发生在外祖父精子减数分裂时[12]。
MPS Ⅱ型的主要临床表现为特殊面容,面容丑陋,身材矮小,关节僵硬,心血管及呼吸系统异常,发育迟缓,智力低下及肝脾肿大。临床根据中枢神经系统是否累及划分为轻型和重型。轻型患者常常于10岁前起病,中枢神经系统无累及,临床症状较轻者的寿命常常无影响。而重型患者常常在1岁6月龄至3岁期间发病,累及中枢神经系统,有智力落后等症状,可在少年期死亡。本例患儿起病于1.5 岁左右,有智力障碍表现,是为重症。
目前MPS 诊断依据尿 GAGs 定性检测,Alder-Reilly bodies 检测,IDS 突变检测,IDS 酶活性检测。而MPS Ⅱ的确诊需要进行IDS 酶活性检测[15],但由于酶活性所用试剂的缺乏,故目前诊断主要是基因诊断为主。
目前MPS Ⅱ型的治疗包括酶替代疗法,即艾杜糖醛酸硫酸酯酶替代疗法,目前已在美国和欧洲批准应用于临床患者中,且酶替代疗法目前在亚洲地区也已经逐渐开展了临床试验,但并不能彻底的治愈MPSⅡ型,且价格昂贵,患者需终身治疗,一般的家庭很难支付高昂的费用。造血多能干细胞细胞移植一般认为在2 岁前实施疗效较好,但由于致死率和病残率问题使该治疗方法仍具有争议性。国内近年报道,采用异基因造血干细胞移植2例MPS Ⅱ型男性患儿,分别于3岁8月龄和2岁11月龄时移植时,移植后疗效显著[16]。基因治疗目前仅限于动物实验及临床研究[17-18]。
尽管依据ACMG 和AMP 的“基因序列变异的解释标准和指南”进行致病性分析显示,尚无法明确该c.820dupG变异位点的致病性,且因为条件限制未对本例患儿行酶学检测,但患儿有MPS Ⅱ型典型的临床表现,且c.820 dupG 变异不属于多态性变化,在人群中出现的概率极其低下(参考数据库1000 Genomes及dbSNP)。故综合考虑基因结果及患者症状及家族史等信息,可基本确认c.820dupG变异位点为患儿及其哥哥的致病性变异。
