七-(2,6-二甲基)-β -环糊精对喜树碱增溶作用的机理
2020-06-04韩东旭李昊原辛士刚张洪波
于 湛, 韩东旭, 李昊原, 辛士刚, 张洪波
(1. 沈阳师范大学 化学化工学院, 沈阳 110034; 2. 沈阳师范大学 实验教学中心, 沈阳 110034)
0 引 言
环糊精(cyclodextrin,CD)是以包含6个(α-)、7个(β-)或8个(γ-)吡喃葡萄糖为基本结构单元,并由α-1,4糖苷键链接而成的环状寡糖[1]。环糊精具有“内腔疏水,外部亲水”的特性,外形呈现上窄下宽、两端开口的中空结构特点。正是由于具有上述结构特点,CD可作为主体与多种无机或有机小分子化合物形成复合物[2]。天然CD特别是β-CD由于具有较强的分子内氢键作用,因此在水中溶解度很小。CD的2-、3-或6-羟基具有一定化学活性,可在一定条件下被其他官能团所取代,生成诸如七-(2,6-二甲基)-β-环糊精(DM-β-CD)、七-(2-羟丙基)-β-环糊精(2HP-β-CD)与6-葡萄糖基-β-环糊精(6Glu-β-CD)等CD衍生物。这些CD衍生物在保留了空腔疏水性的前提下,极大地提高了溶解度与选择性,因此具有良好的应用前景[3-6]。
喜树碱(camptothecin,CPT)是一种由珙桐科植物喜树的果实或根茎表皮中提取的细胞毒性喹啉类生物碱。CPT是被用于早期中医治疗癌症的一种药物,由于其对DNA拓扑异构酶I具有抑制作用[7],成为早期唯一的一种能够作用于DNA拓扑异构酶I的抗肿瘤药物。由于具有抗肿瘤的功效,CPT广泛应用于直肠癌、肠癌、胃癌和白血病等疾病的治疗[8],同时也有一定的副作用,例如呕吐、腹泻[9]及血性膀胱炎等[10]。尽管CPT具有很好的药用前景,但是由于其结构中亲水性极性基团数量较少,在水中的溶解度较低,口服后在人体内的吸收利用率不高。
CD可通过与难溶性小分子药物形成复合物的方式有效提高药物的溶解度[11]。为了提高CPT水溶性,使其能更好地被生物体吸收,本文使用α-CD、β-CD、γ-CD、2HP-β-CD、DM-β-CD与6Glu-β-CD等6种CD作为增溶剂,研究它们对CPT的增溶作用,并通过分子模拟计算推测了增溶机理。

图1 文中所用CD(A)与CPT(B)的化学结构Fig.1 Chemical structures of CDs (A) and CPT (B) used in this work
1 实验部分
1.1 仪器与试剂
傅里叶变换红外光谱仪(NICOLET 380,美国赛默飞),紫外-可见光谱仪(UH5300,日本日立),高速离心机(CT6TA,中国天美)。
CPT购买于西安开来生物制药有限公司,α-CD、β-CD、γ-CD、DM-β-CD、2HP-β-CD和6Glu-β-CD均来自上海源叶生物科技有限公司,本文所使用的溶剂均为分析纯或更高,实验用水为超纯水(25 ℃时电阻率为18.2 MΩ·cm)。
1.2 实验方法
1.2.1 相溶解度法
准确配制50 mL浓度为0.1 mol/L的CD标准溶液待用。准确称量6份质量相同的CPT于15 mL离心管中,每份质量为0.001 0 g,再分别向这6支离心管中加入一定体积CD储液并用水稀释至10.0 mL,使其中CD浓度分别达到0、2.0×10-4、4.0×10-4、6.0×10-4、8.0×10-4和1.0×10-3mol/L。将上述样品在室温下用连续超声30 min后在3 000 rpm条件下离心10 min,取上层清液通过0.22 μm微孔滤膜进行过滤,然后使用紫外-可见光谱分析。
1.2.2 傅里叶变换红外光谱法
以DM-β-CD为例。将CPT、DM-β-CD、DM-β-CD/CPT物理混合物(摩尔比1∶1,研磨30 s)、DM-β-CD/CPT复合物(摩尔比1∶1,研磨30 min)干燥备用。分别取上述4种样品与KBr按1∶100混合于玛瑙研体中,在红外光灯下混合研磨均匀后压片,在波数400~4 000 cm-1范围内检测其红外吸收。
1.2.3 分子模拟计算
α-CD(编号:BAJJAX)、β-CD(编号:ARUXIU)、γ-CD(编号:CYDXPL)的三维结构均来自于剑桥晶体数据库。DM-β-CD、2HP-β-CD与6Glu-β-CD等β-CD衍生物结构是基于β-CD结构使用PyMol软件自行绘制获得的。CPT结构数据来自PubChem网站。CPT与所有CD分子在进行分子模拟计算前,均使用MMFF94方法[12]进行预处理。
本文使用Autodock 4.2软件包进行主客体对接模拟[13]。主客体分子均依照Autodock系统程序默认值进行处理,未改变单键的自由度、非极性氢原子等。分子对接选择在一个126 Å×126 Å×126 Å的立方体格子中进行,格子间隔为默认值0.375 Å,对接计算采用的是拉马克遗传算法(LGA)。为了使每次计算更为充分,以便获得更为准确的计算结果,本文采用较大的运算参数以提高对接计算量,部分参数设置为:最大能量评估值(ga_num_evals)增加至2 500 000,种群数(ga_pop_size)提高至150,循环计算次数(ga_run)提高至300。
本文利用半经验量子化学计算软件MOPAC 2016(V17.279L)对分子对接所获得的结果在半经验水平进行量子化学计算[14],以便于探究复合物形成前后CPT与CD能量变化情况。分别采用PM6-D3H4和PM7方法进行计算,计算参数还包括XYZ、PRECISE、EF及GNOME=0.1,温度为298.15 K。
复合物的复合能ΔEcomplexation依照下述公式计算[15-16]:
ΔEcomplexation=Ecomplex-(Ehost+Eguest) (1)
能量最低的分子对接结果随后导入Schrodinger公司Maestro软件包(2016.04)中,随后使用Desmond分子动力学软件[17]对其进行化学动力学模拟,进而评价分子对接结果的稳定性。
操作顺序如下:首先将能量最低(即得分最高)的分子对接结果导入Maestro中,经过加氢和化学键键级纠错后,再将其置于一个棱长为10 Å的立方体箱子中心,这个箱子中填充满TIP3P模式水分子。由于CPT与CD分子均为电中性并且前述研究中所涉及的水溶液中并无缓冲盐存在,因此这个体系中无需添加任何离子。使用Desmond默认参数进行体系弛豫过程和体系能量的最小化。分子动力学模拟采用NPT系综,温度采用Nose-Hoover耦合方法并设定为300 K,弛豫时间为1 ps,压力采用Martyna-Tobias-Klein方法控制为1.013 25 bar,弛豫时间为2 ps,采用各向同性压强耦合,模拟积分步长设置为5 fs。体系经过2 ns平衡后进行分子动力学模拟,时长为30 ns。
2 结果与讨论
2.1 相溶解度法分析
图2给出了通过监测CPT的λmax处(369.5 nm)吸光度所得到的6种CD存在时CPT的相溶解度曲线。由图可见,在CD浓度为1×10-4~1×10-3mol/L范围内,CPT溶解度S与CD浓度c之间存在良好的线性关系,线性方程及相关系数R2见表1。由图2中曲线可以看出,当浓度为1.0×10-3mol/L时,DM-β-CD具有最大的增溶作用,而γ-CD的增溶作用几乎不随浓度变化,本文推测这可能是γ-CD空腔较大,结合客体能力不强导致。

表1 不同CD/CPT复合物的平衡常数Table 1 The stability constants of CD/CPT inclusion complexes

图2 多种CD存在时CPT的相溶解度曲线Fig.2 Phase solubility curves of CPT in aqueous solution with the existence of various CDs
根据Higuchi和Connors[18]的分类,图2中CPT与各种CD的相溶解度曲线均为AL型,表明CPT和CD之间均形成1∶1型复合物,根据公式K=斜率/(S0×(1-斜率))可计算出复合物的结合常数,其中S0为CPT在水中溶解度,斜率是各条拟合直线斜率。经实验获得CPT在水中溶解度S0为1.000 53×10-5mol·L-1,由此计算得到6种CD/CPT复合物的平衡常数K见表1,可以看出DM-β-CD的K值最大,具有最大的增溶作用。

图3 DM-β -CD/CPT复合物(A)、DM-β -CD/CPT物理混合物(B)、DM-β -CD(C)与CPT(D)的红外光谱图Fig.3 FT-IR spectra of DM-β -CD/CPT inclusion complex(A), DM-β -CD/CPT physical mixture(B), DM-β -CD(C) andCPT(D), respectively
2.2 红外光谱分析
图3为DM-β-CD/CPT复合物、DM-β-CD/CPT物理混合物、DM-β-CD与CPT红外光谱图。通过比对可以看出,1 741 cm-1处羰基吸收峰在物理混合物与复合物谱图中均存在,但是在复合物谱图中强度明显下降,显示复合物中此羰基与DM-β-CD存在相互作用;CPT的1 581 cm-1处吸收峰则在复合物谱图中完全消失,说明CPT的芳香环骨架振动受到严重限制,推测是复合物中CPT复合在DM-β-CD空腔中所致。
2.3 分子模拟计算结果
本文采用分子对接推测DM-β-CD/CPT复合物的结构,300次分子对接后所得的能量最低对接结果示于图4。从图可知CPT分子沿着大口深入DM-β-CD空腔中,芳香环部分几乎完全被包结在DM-β-CD疏水性空腔内,羟基、羰基等官能团位于DM-β-CD大口边缘处,并且分别与DM-β-CD之间形成距离分别为2.3、2.6与3.0 Å的3个氢键。
为了考查分子对接结果的可靠性, 本文还通过半经验计算探究了复合物形成前后主客体分子能量变化情况, 所得结果列于表2。 表2所示的ΔEcomplexation值均小于0可知复合物能量小于主客体能量之和, 即CPT与DM-β-CD复合在热力学上是自发的。无论采用PM6-D3H4还是PM7方法计算, 都可获得相近的结果,说明DM-β-CD/CPT复合物具有很好的热力学稳定性, 验证了分子对接结果的可靠性。

表2 DM-β -CD、CPT与DM-β -CD/CPT的半经验计算结果Table 2 Semiempirical calculation results of DM-β -CD, CPT and DM-β -CD/CPT
本文还通过分子动力学模拟考查了分子对接结果的可靠性和复合物的动力学稳定性。图5为30 ns时间内DM-β-CD/CPT复合物中主、客体相对初始位置RMSD随时间的变化情况。由图可见在分子动力学模拟开始后,体系迅速达到平衡,主、客体中原子位置的变化均在非常小的范围内。在30 ns时间内,DM-β-CD与CPT的平均RMSD分别为1.772 1与0.423 8 Å,标准偏差(SD)均比较小,分别为0.271 3与0.051 93 Å,表明主客体在30 ns时间内主客体均未发生较大的位置改变与形变,说明复合物结构稳定,动力学稳定性强。

图4 DM-β -CD与CPT最优分子对接结果
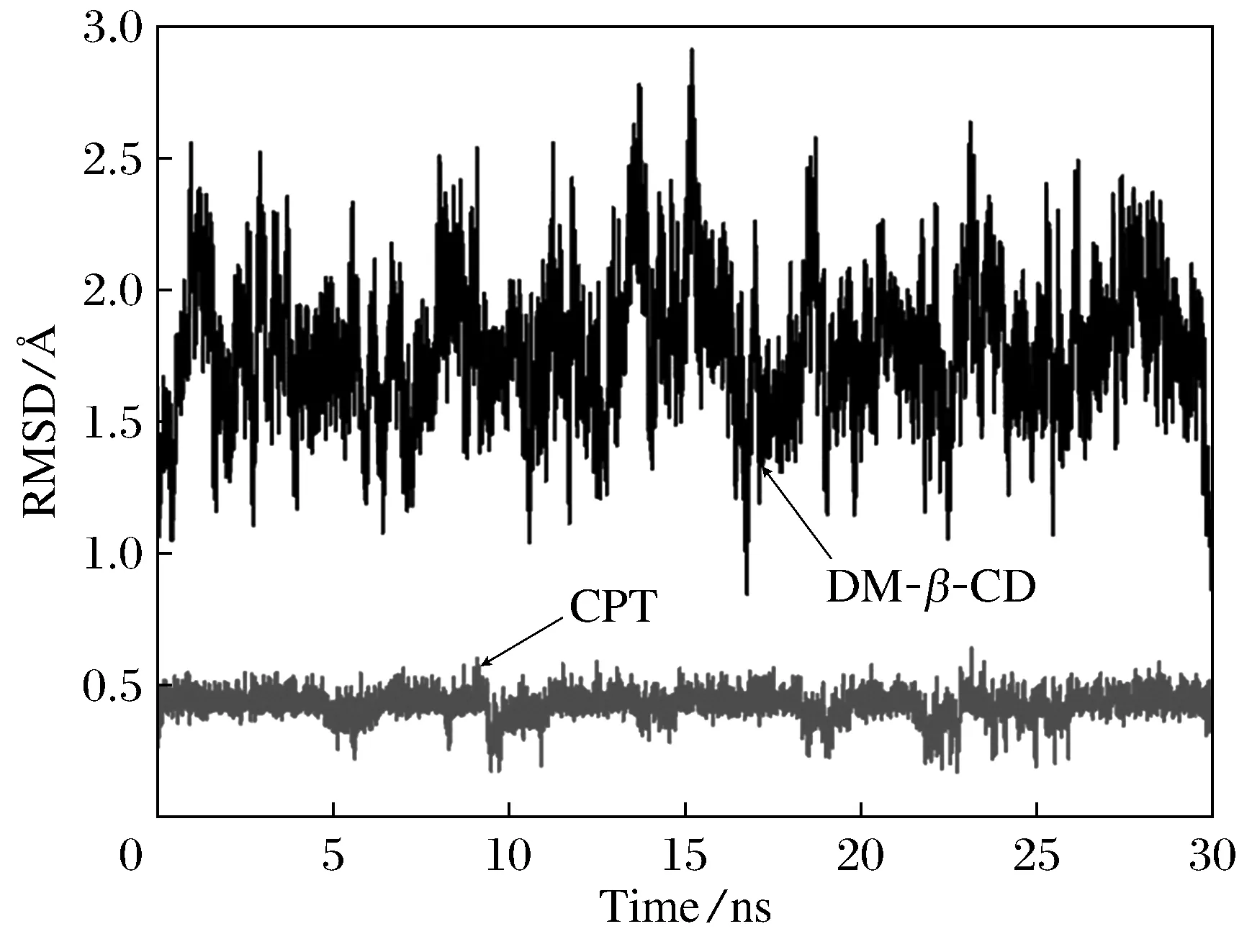
图5 DM-β -CD/CPT复合物中DM-β -CD和CPT相对于初始结构的RMSD随时间变化图
3 结 论
本文采用相溶解度法研究了α-CD、β-CD、γ-CD、DM-β-CD、2HP-β-CD、6Glu-β-CD等6种CD对CPT的增溶,结果表明这些增溶剂均可以与CPT形成1∶1型复合物并具有一定的增溶能力,其中DM-β-CD具有最强的增溶作用。
本文还通过FT-IR法验证了DM-β-CD/CPT复合物的存在与结构特点,并采用分子对接技术推测了复合物的可能结构,结果表明CPT通过进入DM-β-CD空腔并且与端口处氧原子之间形成氢键方式形成复合物。半经验量子化学计算与分子动力学模拟表明DM-β-CD/CPT复合物具有很强的热力学与动力学稳定性。
天然环糊精具有安全无毒、包结能力强的特点,作为药物辅料广泛应用于各种药物制剂研究中。化学修饰可提高环糊精的水溶性并影响其空腔大小与性质,虽然DM-β-CD的空腔体积略小于β-CD,但是具有更好的包结能力,与药物形成的复合物也具有更好的稳定性与水溶性。复合物中主客体之间存在明显的疏水作用与氢键并具有良好的稳定性。本文研究结果表明,DM-β-CD是一种CPT的良好增溶剂,由于其毒性低[19]、增溶效果明显、所形成的复合物稳定性强,在难溶性药物制剂开发与临床应用方面具有很好的潜力。
