基于多反应监测质谱技术的水稻叶片蛋白绝对定量方法
2020-05-30陈铭学杨欢曹赵云马有宁曹珍珍程方民
陈铭学 杨欢 曹赵云 马有宁 曹珍珍 程方民
基于多反应监测质谱技术的水稻叶片蛋白绝对定量方法
陈铭学1,2,#杨欢2,#曹赵云2马有宁2曹珍珍2程方民1,*
(1浙江大学 农业与生物技术学院,杭州 310058;2中国水稻研究所,杭州 310006;#共同第一作者;*通信联系人,E-mail: chengfm@zju.edu.cn)
【】建立水稻叶片蛋白的多反应监测(MRM)质谱绝对定量方法。水稻叶片蛋白经含1.0%十二烷基硫酸钠(SDS)的磷酸盐(PBS)缓冲液提取,丙酮沉淀除杂纯化、胰蛋白酶消化,酶解液经液相色谱分离,MRM质谱监测,外标法定量。向提取缓冲液中加入1.0% SDS可增强水稻叶片中16种靶蛋白和总蛋白质的提取效果;不同有机试剂处理,总蛋白质沉淀量存在显著差异(<0.05),沉淀能力从强到弱依次为乙腈>丙酮>异丙醇>甲醇>乙醇;对于16种目标蛋白,总体以丙酮沉淀效果最好,其次是异丙醇和乙腈,甲醇和乙醇效果较差。该方法线性范围均达到3个数量级,定量限为0.1~2.5 nmol/L,灌浆期16种水稻叶片蛋白质含量为6.0~2818.1 μg/g,相对标准偏差均小于14%。SDS可显著提高水稻叶片蛋白提取效果,采用丙酮或乙腈可获得较好的蛋白沉淀效果,但不同蛋白质略有差异。结合MRM质谱监测技术可实现水稻叶片蛋白的绝对定量,方法线性范围宽、敏度高、重复性好。
水稻叶片;蛋白质;多反应监测;SOD法;绝对定量
生物体内蛋白质丰度的动态变化对各种生命过程有着重要影响[1],准确测定不同状态下复杂生物体内蛋白质表达量及量的变化,是定量蛋白质组学研究的重要内容[2]。近年来,高分辨率、高扫描速度生物质谱技术的进步,推动了蛋白绝对定量技术的发展。其中,多反应监测(multiple reaction monitoring, MRM)质谱具有选择性强、灵敏度高、线性宽等技术优势[3],被视为靶向蛋白绝对定量的重要手段,并已应用于特异性生物标志物筛的选验证[4-5]、食物过敏源的痕量检测[6-7]及植物中重要功能蛋白质的定量分析[8]等领域。
尽管MRM质谱技术具有诸多技术优势,但研究发现,目标蛋白提取效果、非蛋白组分干扰等因素均会严重影响方法准确度和重复性[9],这给现有的蛋白样本制备手段带来巨大挑战。由于生物基质极为复杂,尤其是植物组织中存在大量蛋白酶、厚壁细胞和次生代谢化合物(如酚类和色素物质等)[10],严重影响蛋白的提取率和质谱分析。现有的植物蛋白质提取方法如酚法、三氯乙酸(trichloroacetic acid, TCA)-丙酮沉淀法等均存在蛋白质提取步骤繁琐,耗时长,沉淀后蛋白复溶不完全等问题[11-12],难以满足定量分析的要求。此外,传统样品制备过程中的内源共提物和外源变性剂等对质谱的离子碎片采集带来干扰,进而对方法准确度和精密度造成不利影响[13]。因此,建立高效的蛋白样品制备方法成为MRM质谱蛋白定量技术的核心内容。
水稻(L.)是最重要的粮食作物之一,准确测定不同生理或病理条件下蛋白质表达量的变化,对于阐明自身防御系统的分子机制,增强作物抗逆能力及提高籽粒产量与品质等具有重要意义[8,14]。截至目前,有关水稻蛋白质的绝对定量技术却鲜有报道。本研究根据特异性肽段的疏水性、等电点、肽段长度及在色谱中的保留行为等特征,选择16种典型肽段对应的水稻叶片蛋白为研究对象[15],采用表面活性剂辅助沉淀/颗粒酶解(surfactant aided precipitation/on pellet digestion, SOD)蛋白提取、有机试剂沉淀除杂等系列技术,建立了适用于水稻叶片蛋白的绝对定量方法。该方法的建立,将为水稻MRM质谱定量蛋白组学研究提供技术参考。
1 材料与方法
1.1 仪器设备与试剂
LC-20ADXR 液相色谱仪(日本岛津公司);AB SCIEX QTRAP 5500质谱仪(美国AB Sciex公司);Poroshell 120 EC-C18色谱柱(150 mm×2.1 mm, 2.7 μm, 美国Agilent公司);UV-2600紫外分光光度计(日本岛津公司);SD-C18固相萃取小柱(美国3M公司);Milli-Q超纯水系统(美国Millipore公司)。
乙腈、甲醇、丙酮、乙醇和异丙醇(德国Merck公司);十二烷基硫酸钠(sodium dodecyl sulfate, SDS)残留试剂盒、20×PBS缓冲液、牛血清蛋白和BCA蛋白质定量试剂盒(上海Sangon公司);乙二胺四乙酸、碳酸氢铵(NH4HCO3,Acros Organics公司);甲酸(美国Fluka公司);蔗糖(纯度≥99%,阿拉丁试剂有限公司);SDS、碘乙酰胺(iodoacetamide, IAA)、胰蛋白酶、尿素、蛋白酶抑制剂(美国Sigma公司);3-[3-(胆酰胺丙基)二甲氨基]丙磺酸内盐{3-[3-(cholamidopropyl)dimethylamino]propanesulfonic acid inner salt, CHAPS}、二硫苏糖醇(dithiothreitol, DTT,Promega公司),以上试剂均为分析纯或色谱纯。16种特征肽段标准品(纯度≥95%)均由上海强耀生物科技公司合成。
1.2 水稻叶片总蛋白质样品制备及酶解
1.2.1 SOD法
供试水稻品种为9311,于2018年5月10日播种并按大田常规水肥管理,待水稻生长至灌浆期,取剑叶叶片,剪去中脉后置于−80℃冰箱冻藏。称取0.5 g经液氮研磨后的水稻叶片样品置于50 mL离心管中,加入5 mL含1.0% SDS的PBS缓冲液(100 mmol/L, pH 7.4),冰浴中高速匀浆超声30 s,于4℃、10 000×条件下离心30 min。取上清液分别加入50 μL 1 mol/L DTT使其终浓度为10 mmol/L,于56 ℃条件下反应30 min;加入125 μL 1 mol/L IAA使其终浓度为25 mmol/L,于37 ℃条件下避光孵化30 min。加入等体积(5 mL)的预冷丙酮混匀,再加入5倍体积的预冷丙酮剧烈震荡,置于−20 ℃条件下静置3 h。取出于4 ℃、10 000×离心45 min,弃去上清液。加入等体积的丙酮/水混合液(丙酮∶水=6∶1)清洗沉淀一次,离心30 min后弃去上清液在空气中自然晾干。用1 mL 50 mmol/L NH4HCO3(pH 8.0)充分溶解蛋白质,于4℃、10 000×条件下离心30 min,取上清液采用BCA试剂盒测定蛋白质浓度。酶解参照Kilambi等[12]的方法。
1.2.2 酚法
参照高欢欢等[15]报道的方法进行。
1.2.3 TCA/丙酮沉淀法
参照Ippoushi等[16]报道的方法进行。
1.3 水稻叶片蛋白质酶解液净化
按Wisniewski等[17]的方法将上述1.2中3种方法获得的酶解样品经SD-C18柱吸附净化。具体净化步骤如下:依次用1 mL甲醇、0.5 mL 70%乙腈水溶液(含0.1%甲酸)和0.5 mL 0.1%甲酸溶液活化平衡SD-C18固相萃取小柱;将酶解液装载于小柱中,在4 ℃、300×下离心3 min;加入0.5 mL 0.1%甲酸溶液冲洗小柱后,用0.5 mL 70%乙腈水溶液(含0.1%甲酸)洗脱,并重复洗脱一次。将两次洗脱液混匀过0.22 μm滤膜后上LC-MS/MS分析。
Fig. 1. Total ion chromatogram of 16 kinds of signature peptides.
1.4 仪器采集条件
液相色谱条件:流动相分别为0.1%甲酸溶液(A)和乙腈(B),洗脱梯度为:0~30 min,5%~90% B;30~35 min,90% B;35~35.1 min, 90%~5% B;35.1 ~40 min,5% B。流速为0.2 mL/min,柱温为40℃。
质谱条件:电喷雾正离子模式(ESI+);气帘气压力为25 psi;离子源温度为500 ℃;Gas1和Gas2的压力均为30 psi;MRM模式采集。
1.5 数据分析
QTRAP 5500数据分析在Ananlyst 1.6.2软件处理系统上进行。采用SPSS Statistics 19.0软件(Chicago, Illinois, USA)进行对数据进行统计分析,统计学显著性水平为95%(<0.05),采用OriginPro 8.5(美国OriginLab公司)作图。
2 结果与分析
2.1 MRM采集方法的建立
MRM定量蛋白的技术核心是对靶标蛋白酶解产生的特异性肽段进行质谱监测,但肽段产生的碎片离子通常比药物或小分子代谢物更为复杂,需选择优化合适的离子对和质谱采集参数[18]。为此,实验取16种特征肽段的单一母液,用50%乙腈水溶液(含0.1%甲酸)配制成浓度为100.0 μg/L的混合标准溶液。将该溶液引入质谱,利用Skyline软件对16种特征肽段的质谱参数(去簇电压、碰撞池出口电压和入口电压)进行预测和优化,并获得相应42组离子对的MRM采集参数。
实验以0.1%甲酸水溶液和乙腈分别为A相和B相,优化洗脱梯度。结果表明,16种特征肽段均在反相色谱柱上获得较好的保留和良好的分离效果,且单次运行时间小于30 min(16种特征肽段的总离子流见图1)。通过连续50次进样发现,各肽段色谱峰面积的相对标准偏差(RSD)均在10%以内,保留时间RSD均小于1.1%。由此说明,该MRM采集方法具有分离效果好、分析速度快、重复性好等特点,能满足同时分析16种目标蛋白质的技术要求。
将16种特征肽段单储备液用5%乙腈水溶液(含0.1%甲酸)配制成5个不同浓度水平的混合标准溶液,每个浓度标液重复测定3次,外标法定量。分别以16种特征肽段峰面积()对质量浓度(, μg/L)绘制标准曲线。由表1可知,各肽段线性关系良好(相关系数均大于0.99),线性范围均达到3个数量级。以信噪比(S/N)分别≥3∶1和≥10∶1计算得到的浓度表示仪器检出限(limits of detection, LOD)和定量限(limits of quantification, LOQ)。16种特征肽段的仪器LOD和LOQ范围分别为0.03~0.8 nmol/L和0.1~2.5 nmol/L,可用于水稻叶片中16种功能蛋白质的定量分析。
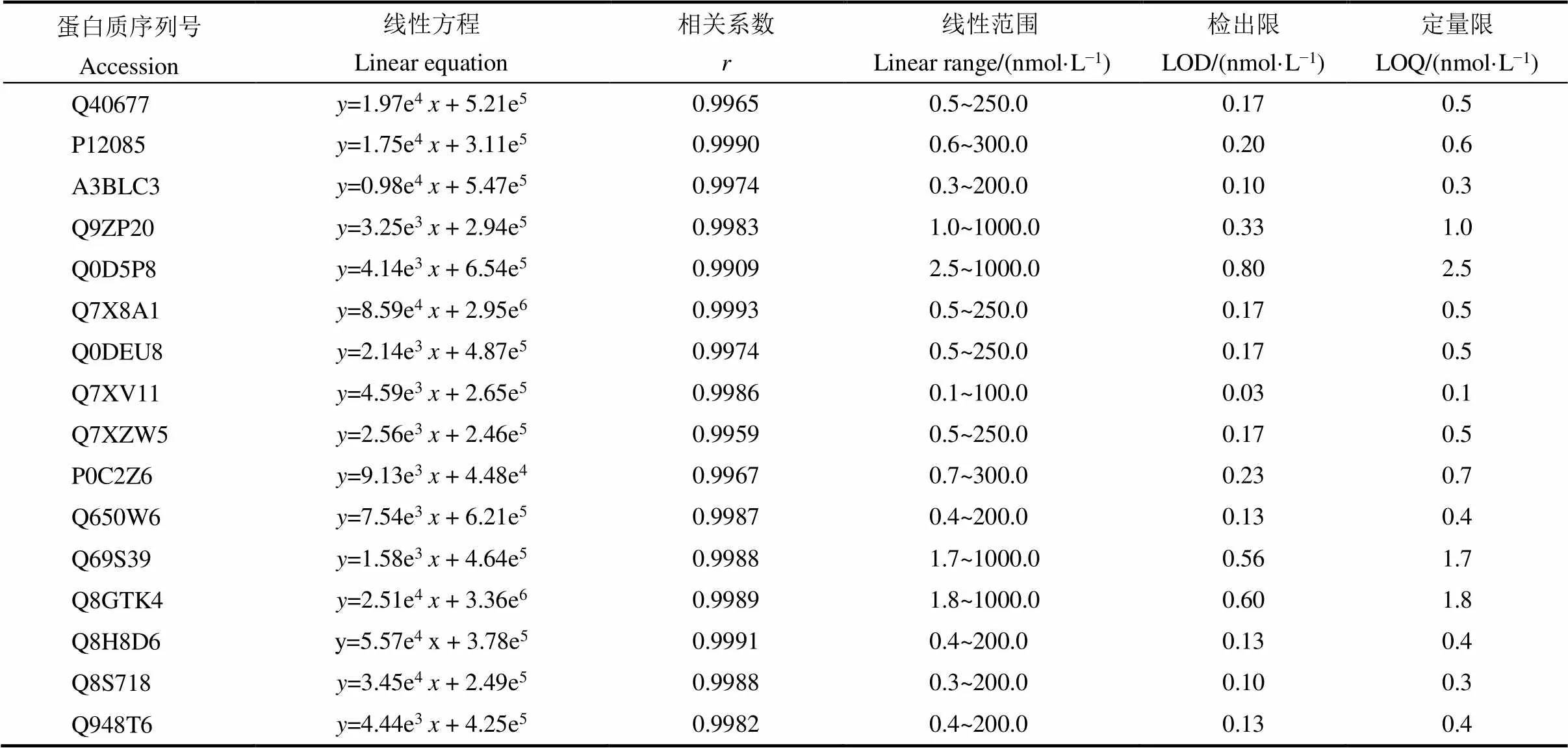
表1 16种特征肽段的线性方程、相关系数及仪器检出限与定量限
2.2 蛋白质SOD提取方法的建立
2.2.1 表面活性剂的选择
表面活性剂可使内源性蛋白质酶抑制剂失效,促进样品中蛋白质的变性、还原和烷基化,有助于去除影响胰蛋白酶水解和LC-MS分析的小分子杂质(如磷脂)[19]。实验研究了不同SDS浓度(0%、0.1%、0.5%、1.0%、2.0%、4.0%)对16种目标蛋白质和总蛋白质提取量的影响。结果(图2)表明,Q0D5P8等13种目标蛋白提取量随着SDS浓度的增加呈先上升后降低的变化趋势,在SDS含量为1.0%或2.0%时达到峰值;总蛋白随着提取液中SDS浓度的增加而提高,但当表面活性浓度大于1.0%时,总蛋白浓度趋于饱和。因此,含1.0%SDS的PBS缓冲液(100 mmol/L, pH 7.4)可获得较高浓度的蛋白质。
此外,我们还考查了其他表面活性剂对水稻叶片蛋白的提取效果。首先,向PBS提取缓冲液(100 mmol/L, pH 7.4)中加入相同浓度(1.0%)的CHAPS和SDS,并以PBS提取缓冲液为对照,采用已建立的MRM质谱法对16种目标蛋白提取量进行比较。结果表明,使用CHAPS和SDS均有利于促进目标蛋白的提取,但使用后者的效果更好。其中,使用SDS获得的12种目标蛋白提取浓度显著高于CHAPS和对照(<0.05),增幅分别达14.2%~80.7%和23.4%~138.6%,其余4种蛋白提取量则无明显差异。与上述16种目标蛋白实验结果类似,对3种提取液获得的总蛋白考查结果表明,提取液中加入SDS获得的蛋白总量最高为9.9±0.7 mg/mL,分别较CHAPS和对照高59.6%和83.3%。上述结果表明,SDS是促进水稻叶片蛋白提取的有效手段。
2.2.2 不同种类有机试剂的沉淀效果比较
使用有机试剂不仅可去除粗蛋白中可溶性盐、脂类、核酸等杂质,也能有效去除提取步骤引入的SDS[20]。蛋白质的沉淀效果不仅受蛋白质种类、分子量的影响,也与沉淀试剂种类密切相关。因此,实验考查了乙腈、乙醇、丙酮、甲醇和异丙醇5种有机溶剂对蛋白质的沉淀效果和SDS的去除作用。
实验结果表明,5种有机试剂处理,总蛋白质沉淀量从高到低依次为乙腈>丙酮>异丙醇>甲醇>乙醇(<0.05)。但从图3可以看出,11种目标蛋白采用丙酮沉淀效果最好,少数(如Q9ZP20、Q0D5P8、Q8H8D6)采用异丙醇或乙腈可获得最佳沉淀效果。但总体而言,以丙酮效果最好,其次是乙腈和异丙醇,甲醇和乙醇则效果较差。这与不同有机试剂沉淀总蛋白效果略有差异,说明沉淀试剂对蛋白沉淀效果具有一定选择性。
Bereman等[13]研究表明,过高浓度的SDS会抑制胰蛋白酶活性,干扰RPLC的分离和MS分析。因此,SDS残留量可作为粗蛋白沉淀除杂的一项重要指标。实验结果显示,采用上述5种有机试剂对SDS的去除率均可达94.2%以上,均能保证样品中SDS浓度低于0.01%,符合HPLC和LC/MS分析条件[21]。综上,实验选择丙酮作为16种目标蛋白的沉淀剂。

图中数据为均值±标准差(n=3);不同字母表示在0.05水平上有显著性差异,而具相同字母表示无显著性差异。纵坐标为蛋白质相对提取百分比,以未加SDS的提取液对应的各目标蛋白提取量为100%计算。
Fig. 2. Influence of different concentrations of SDS in extraction buffer on the extraction effects of 16 kinds of proteins in rice leaves.
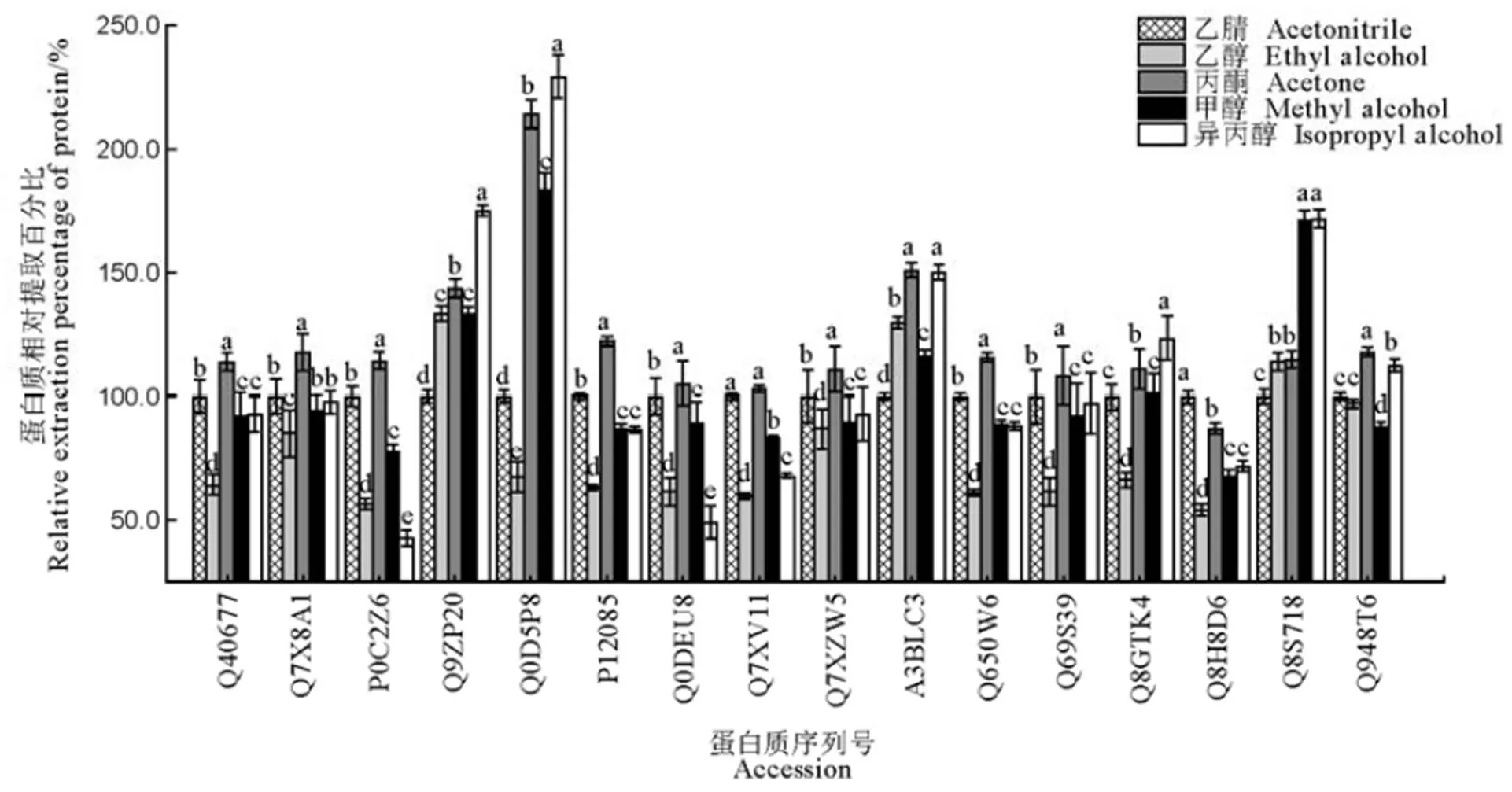
图中数据为均值±标准差(n=3),不同字母表示在0.05水平上有显著性差异,而具相同字母表示无显著性差异。纵坐标为蛋白质相对提取百分比,以乙腈沉淀对应的各目标蛋白提取量为100%计。
Fig. 3. Comparison of precipitation effects of 16 kinds of proteins in rice leaves among five different organic reagents.
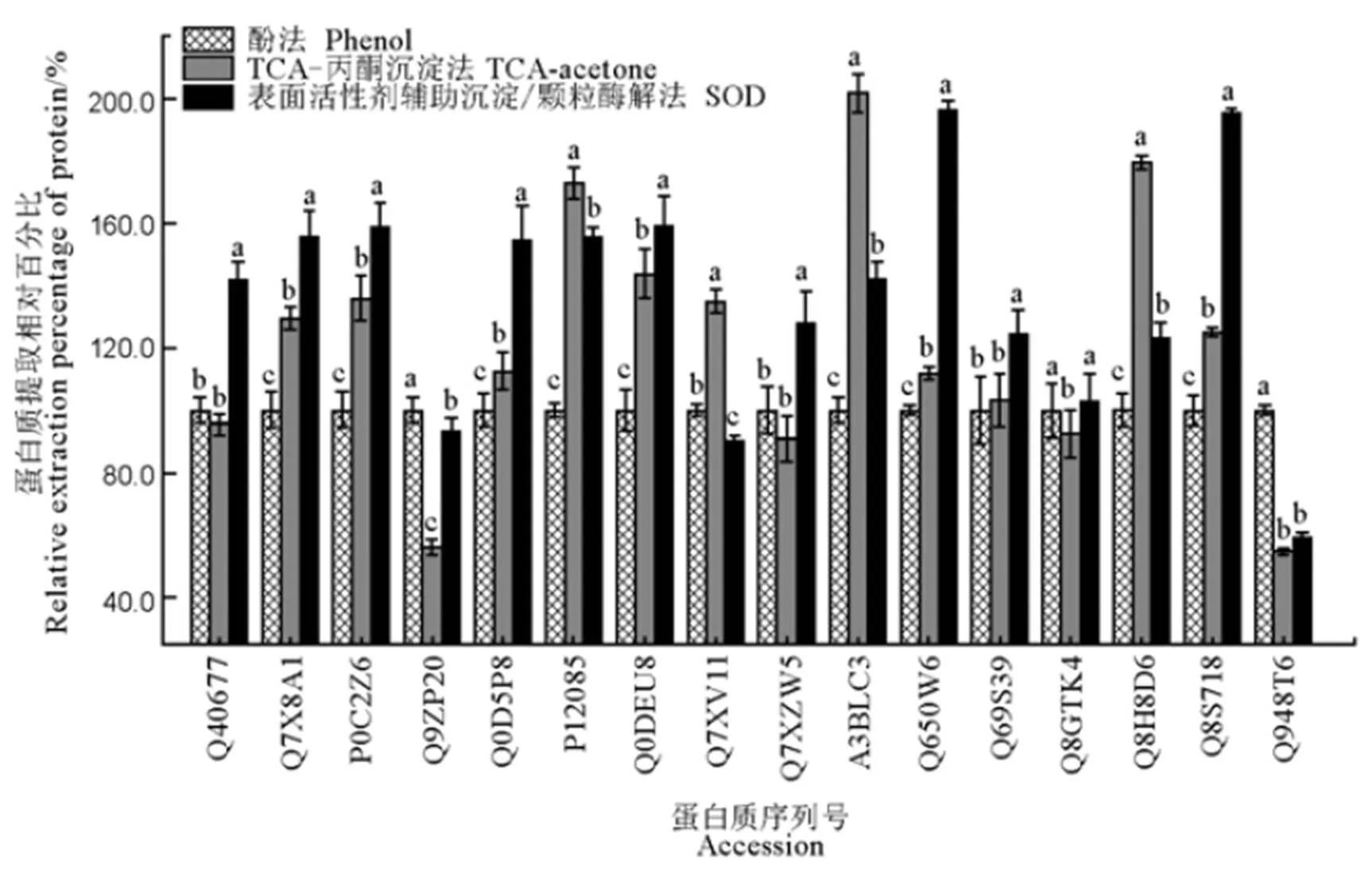
图中数据为均值±标准差(n=3),不同字母表示在0.05水平上有显著性差异,而具相同字母表示无显著性差异。纵坐标为蛋白质相对提取量,以酚法对应的各目标蛋白提取量为100%计。
Fig. 4. Comparison of extraction effects of 16 proteins in rice leaves by SOD, phenol and TCA-acetone precipitation methods.
2.2.3 SOD法与酚法、TCA-丙酮沉淀法比较
将已建立的SOD法与传统酚法和TCA-丙酮沉淀法对16种目标蛋白质和总蛋白的提取效果进行比较(图4)。由图4可以看出,采用SOD法对Q40667等9种蛋白的提取量均显著高于另两种方法(<0.05),对应提取量分别比酚法和TCA-丙酮沉淀法高24.7%~96.5%和15.6%~84.7%;TCA-丙酮沉淀法仅对P12085等4种蛋白质的提取量显著高于SOD法和酚法(<0.05);相对而言,酚法的提取效果最差。总蛋白提取量显示,SOD法提取的总蛋白与酚法无显著差异,但均显著高于TCA-丙酮沉淀法(<0.05)。综合考虑,与酚法和TCA-丙酮沉淀法相比,SOD法更适用于水稻叶片中16种重要功能蛋白质定量分析的样品制备。
2.3 方法应用
按上述1.2.1 SOD法重复制备蛋白质样品(=6),采用LC-MS/MS和外标法测定16种水稻叶片蛋白含量。结果如表2所示,灌浆期16种叶片蛋白质的含量范围在6.0~2818.1 μg/g,相对标准偏差均在13.9%以下。其中,Q69S39含量最高(2818.1±219.9 μg/g),该蛋白是位于叶绿体类囊体膜上,参与光合作用的细胞色素b6-f复合物重要组分,但还未见对其绝对含量的相关报道。因此,本实验所建立的SOD法能有效地提取水稻叶片中蛋白质,并结合MRM质谱技术和外标法,实现水稻灌浆期16种叶片蛋白质的绝对定量。

表2 SOD法测定水稻16种叶片蛋白质绝对含量(n=6)
RSD, Relative standard deviation.
3 讨论
SDS阴离子变性剂一方面破坏蛋白质的二级和三级结构,提高蛋白质溶解度和酶解,在蛋白质(特别是膜蛋白)测定和蛋白质组的全面表征中有所应用[22]。另一方面,Ilavenil等[23]研究表明浓度过高(>1.0%)时也会抑制胰蛋白酶活性,过量的SDS会产生DS−,抑制离子电离,影响质谱分析结果。实验对提取液中SDS浓度优化结果发现,SDS浓度在1.0%以内,16种目标蛋白和总蛋白提取量随浓度增大而增加,但超过1.0%后逐渐趋于饱和甚至呈下降趋势。该结果与An等[19]研究SDS对血浆中治疗蛋白提取效率影响的结果一致。因此,SDS可作为一种通用技术,用于生物组织中蛋白质的提取。
有机试剂一方面能降低溶液的电解常数,导致溶解度下降;另一方面与水作用,破坏蛋白颗粒的水化膜,使大部分蛋白质变性聚集沉淀。实验研究不同种类有机试剂对水稻叶片蛋白的沉淀效果发现,乙腈、甲醇、丙酮等5种有机试剂对总蛋白和16种目标蛋白沉淀效果存在差异。总蛋白质沉淀量从高到低依次为乙腈>丙酮>异丙醇>甲醇>乙醇,但绝大多数目标蛋白则采用丙酮沉淀可获得最高丰度,少数(如Q9ZP20、Q0D5P8、Q8H8D6)采用异丙醇或乙腈可获得最佳沉淀效果。这种现象说明不同沉淀剂对蛋白质具有一定的选择性。如Ouyang[24]比较多种试剂对血浆中治疗蛋白的沉淀效果发现,尽管乙腈沉淀可得到最大蛋白浓度,但甲醇沉淀更利于目标蛋白后续复溶和酶解研究。对于烟曲霉的分泌蛋白而言,氯仿/甲醇沉淀后能获得最高的蛋白质回收率[25],但海马等中药材中的功能蛋白质在丙酮沉淀下能获得较为全面的蛋白质表达谱[26]。因此,针对基质背景各异和种类特性不同的蛋白质,需合理选择有机试剂沉淀以达到高效提取。
此外,丙酮沉淀既能浓缩蛋白,也可以除去粗提液中的可溶性杂质,起到纯化蛋白的作用。尽管去垢小柱、FASP和MOFs材料等方法被用于去除样品中残留的SDS,但存在成本高,操作步骤繁琐等问题。Santa等[20]采用丙酮沉淀去除提取液中SDS,有效避免残留SDS对酶解和定量分析的干扰,提高了蛋白质的回收率和重现性。
复杂组分共存干扰物和待测蛋白丰度差异大,一直是影响生物体中蛋白准确定量的关键因素。尽管人们尝试多种方法,如提高混合组分分离,去除干扰物等,但对低丰度蛋白质的检测灵敏度和精密度仍不能满足分析的要求。MRM质谱技术则通过特征肽段离子对的选择性监测,降低了复杂组分的背景信号干扰,从而提高了对低丰度靶标蛋白的分析灵敏度,延伸了定量的动态范围。如Rezeli等[27]采用MRM技术对人体血浆中87个心血管等疾病相关蛋白标志物进行定量检测,最大线性动态范围达4到5个数量级,定量结果精密度RSD小于4.7%。此外,Hoofnagle等[28]从方法验证学上,对MRM技术和传统的免疫分析法进行系统比较,结果发现,两者的分析结果相关性达到0.61~0.96,并提出MRM技术可作为传统蛋白定量的替代技术,具有很大的应用价值。本研究结果表明,尽管样品基质较为复杂,但16种目标蛋白的线性范围仍超过2个数量级。此外,虽实际样品中各蛋白丰度差异也非常大(各蛋白含量水平为6.0~2818.1 μg/g),但仍可获得良好的重复性,即各蛋白的分析精密度均小于13.9%。
目前基于MRM质谱技术的蛋白定量方法主要应用于动物组织或细胞中蛋白标志物分析,在植物蛋白组学研究应用较少,其原因主要是缺乏标准化操作流程。此外,该技术需要较高的专业技能、分析成本等也是阻碍其快速发展的重要因素。本研究以水稻叶片中相关蛋白为研究对象,采用SOD蛋白制备方法和MRM质谱分析技术,建立一种通用的目标蛋白绝对定量技术流程,以期促进该分析技术在植物蛋白组学中的推广和运用。
[1] 钱小红. 定量蛋白质组学分析方法[J]. 色谱, 2013, 31(8): 719-723.
Qian X H. Quantitative proteomics analysis[J]., 2013, 31(8): 719-723. (in Chinese with English abstract)
[2] 齐林玉, 姜淼, 郭艳, 王迎, 陈平. 质谱MRM技术结合稳定同位素标记法的应用进展[J]. 生命科学研究, 2015, 19(5): 444-451.
Qi L Y, Jiang M, Guo Y, Wang Y, Chen P. Progresses on application of MS MRM technology combining with stable isotope labeling[J]., 2015, 19(5): 444-451. (in Chinese with English abstract)
[3] Veronika V, Zdenek S. A review on mass spectrometry-based quantitative proteomics: Targeted and data independent acquisition[J]., 2017, 964: 7-23.
[4] 王素兰, 高华萍, 张菁, 叶翔. 基于稳定同位素标记和平行反应监测的蛋白质组学定量技术用于肝癌生物标志物的筛选和验证[J]. 色谱, 2017, 35(9): 934-940.
Wang S L, Gao H P, Zhang J, Ye X. Stable isotope labeling and parallel reaction monitoring-based proteomic quantification for biomarker screening and validation of hepatocellular carcinoma[J]., 2017, 35(9): 934-940. (in Chinese with English abstract)
[5] Chen Y T, Chen H W, Wu C F, Chu L J, Chiang W F, Wu C C, Yu J S, Tsai C H, Liang K H, ChangY S, Wu M , Ou Yang W T. Development of a multiplexed liquid chromatography multiple-reaction-monitoring mass spectrometry (LC-MRM/MS) method for evaluation of salivary proteins as oral cancer biomarkers[J]., 2017, 16(5): 799-811.
[6] Zhang J, Lai S, Cai Z, Chen Q, Huang B, Ren Y. Determination of bovine lactoferrin in dairy products by ultra-high performance liquid chromatography-tandem mass spectrometry based on tryptic signature peptides employing an isotope-labeled winged peptide as internal standard[J]., 2014, 4(829): 33-39.
[7] Planque M, Arnould T, Delahaut P, Renardb P, Dieub M, Gillard N. Development of a strategy for the quantification of food allergens in several food products by mass spectrometry in a routine laboratory[J]., 2019, 274: 35-45.
[8] Rautengarten C, Ebert B, Heazlewood J L. Absolute quantitation of in vitro expressed plant membrane proteins by targeted proteomics (MRM) for the determination of kinetic parameters[J].,2018, 1696: 217-234.
[9] Gong C, Zheng N, Zeng J, Aubry A F, Arnold M E. Post-pellet-digestion precipitation and solid phase extraction: A practical and efficient workflow to extract surrogate peptides for ultra-high performance liquid chromatography-tandem mass spectrometry bioanalysis of a therapeutic antibody in the low ng/mL range[J]., 2015, 1424: 27-36.
[10] Wu X, Xiong E, Wang W, Scali M, Cresti M. Universal sample preparation method integrating trichloroacetic acid/acetone precipitation with phenol extraction for crop proteomic analysis[J]., 2014, 9(2): 362-374.
[11] Luís I M, Alexandre B M, Oliveira M M, Abreu I A. Selection of an appropriate protein extraction method to study the phosphoproteome of maize photosynthetic tissue[J/OL]., 2016, 11(10): e0164387.
[12] Kilambi H V, Manda K, Sanivarapu H, Maurya V K, Sharma R, Sreelakshmi Y. Shotgun proteomics of tomato fruits: Evaluation, optimization and validation of sample preparation methods and mass spectrometric parameters [J/OL]., 2016, 7: 969.
[13] Bereman M S, Egertson J D, MacCoss M J. Comparison between procedures using SDS for shotgun proteomic analyses of complex samples[J]., 2011, 11(14): 2931-5.
[14] Yang L, Ji J, Harris-Shultz K R, Wang H, Wang H, Abd-Allah E F, Luo Y, Hu X Y. The dynamic changes of the plasma membrane proteins and the protective roles of nitric oxide in rice subjected to heavy metal cadmium stress[J/OL]., 2016, 7: 190.
[15] 高欢欢, 马有宁, 林晓燕, 柴爽爽, 秦美玲, 张涵彤, 何巧, 陈铭学. 亲水作用-反相二维液相色谱串联质谱法鉴定水稻蛋白质[J]. 分析化学, 2018, 46(5): 650-657.
Gao H H, Ma Y N, Lin X Y, Chai S S, Qin M L, Zhang H T, He Q, ChenM X. Development of two-dimensional liquid chromatography coupled with tandem mass spectrometry for identification of extracted proteins of rice leaves: Hydrophilic interaction-reversed-phase approach[J]., 2018, 46(5): 650-657. (in Chinese with English abstract)
[16] Ippoushi K, Sasanuma M, Oike H, Kobori M, Maeda-Yamamoto M. Absolute quantification of protein NP24 in tomato fruit by liquid chromatography/tandem mass spectrometry using stable isotope-labelled tryptic peptide standard[J]., 2015, 173: 238-242.
[17] Wisniewski J R, Zouqman A, Nagaraj N, Mann M. Universal sample preparation method for proteome analysis[J]., 2009, 6(5): 359-362.
[18] Bru-Martínez R, Martínez-Márquez A, Morante-Carriel J, Sellés-Marchart S, Martínez-Esteso M J, Pineda-Lucas J L, Luque I. Targeted quantification of isoforms of a thylakoid-bound protein: MRM method development[J]., 2018, 1696: 147-162.
[19] An B, Zhang M, Johnson R W, Qu J. Surfactant-aided precipitation/on-pellet-digestion (SOD) procedure provides robust and rapid sample preparation for reproducible, accurate and sensitive LC/MS quantification of therapeutic protein in plasma and tissues [J]., 2015, 87(7): 4023-4029.
[20] Santa C, Anjo S I, Manadas B. Protein precipitation of diluted samples in SDS-containing buffer with acetone leads to higher protein recovery and reproducibility in comparison with TCA/acetone approach[J].2016, 16(13): 1847-1851.
[21] Botelho D, Wall M J, Vieira D B, Fitzsimmons S, Liu F, Doucette A. Top-down and bottom-up proteomics of SDS containing solutions following mass-based separation[J].2010, 9(6): 2863-2870.
[22] Zhang N, Chen R, Young N, Wishart D, Winter P, Weiner J H, Li L. Comparison of SDS and methanol-assisted protein solubilization and digestion methods formembrane proteome analysis by 2D LC-MS/MS[J]., 2007, 7(4): 484-493.
[23] Ilavenil S, Al-Dhabi N A, Srigopalram S, Kim Y O, Agastian P, BaaruKi R, Choon Choi K C, Arasu M V, Park C G, Park K H. Removal of SDS from biological protein digests for proteomic analysis by mass spectrometry[J]., 2016, 14(1): 11.
[24] Ouyang Z, Furlong M T, Wu S, Sleczka B, Tamura J, Wang H, Suchard S, Suri A, Olah T, Tymiak A, Jemal M. Pellet digestion: A simple and efficient sample preparation technique for LC-MS/MS quantification of large therapeutic proteinsin plasma[J]., 2012, 4(1): 17-28.
[25] Srivastava A K. A Comparison of protein extraction methods using organic solvents for secretome of aspergillus fumigatus strain (MTCC 1811).2015, 4(6): 2277-8179.
[26] 张冬梅, 马慧萍, 贾正平. 纳升级反相液相色谱串联质谱法分析海马蛋白质组[J]. 药物分析杂志, 2018, 38(1): 118-123.
Zhang D M, Ma H P, Jia Z P. Proteomic analysis of Hippocampus using nanoflow reversed phase liquid chromatography-tandem mass spectrometry[J]., 2018, 38(1): 118-123. (in Chinese with English abstract)
[27] Rezeli M, Sjödin K, Lindberg H, Gidlöf O, Lindahl B, Jernberg T, Spaak J, Erlinge D, Marko-Varga G. Quantitation of 87 proteins by nLC-MRM/MS in human plasma: workflow for large-scale analysis of biobank samples[J]., 2017, 16(19): 3242-3254.
[28] Hoofnagle A N, Becker J O, Oda M N, Cavigiolio G, Mayer P, Vaisar T. Multiple-reaction monitoring-mass spectrometric assays can accurately measure the relative protein abundance in complex mixture[J].2012, 58(4): 777-781.
Absolute Quantification of Proteins in Rice Leaf Based on Multiple Reaction Monitoring Mass Spectrometry
CHEN Mingxue1,2,#, YANG Huan2,#, CAO Zhaoyun2, MA Youning2, CAO Zhenzhen2, CHENG Fangmin1,*
(College of Agriculture and Biotechnology,,,;China National Rice Research Institute,,;These authors contributed equally to this work;Corresponding author,:)
【】An absolute quantitative method for proteins in rice leaf based on multiple reaction monitoring (MRM) mass spectrometry was established.【】The proteins in rice leaf were extracted with PBS buffer containing 1.0% sodium dodecyl sulfate(SDS), purified by acetone precipitation, and then digested by Trypsin. The enzymatic hydrolysate was separated by liquid chromatography, monitored by MRM mass spectrometry and quantified by external standard method.【】The addition of 1.0% SDS to the extraction buffer enhanced the extraction effect of 16 kinds of target proteins and total proteins in rice leaf. Total proteins precipitation with several solvents varied significantly (<0.05), and the precipitation ability from strong to weak was acetonitrile>acetone>isopropanol>methanol>ethanol. Acetone performed best in the precipitation effect of 16 kinds of target proteins, followed by isopropanol and acetonitrile, and methanol and ethanol represented the worst precipitants. The relative standard deviations of the method were all less than 14% in three orders of magnitude range, with the quantification limit from 0.1 to 2.5 nmol/L, and the contents of 16 kinds of proteins in rice leaf during the grain filling stage ranged from 6.0 to 2818.1 μg/g.【】SDS could significantly improve the extraction effect of proteins in rice leaf. Acetone and acetonitrile addition generated excellent precipitation effects of proteins, with slight difference in various proteins. Combined with MRM mass spectrometry technology, the absolute quantification method for proteins in rice leaf is featured by wide-linear range, high sensitivity and good repeatability.
rice leaf; protein; multiple reaction monitoring; SOD method; absolute quantification
S511.01
A
1001-7216(2020)03-0278-09
10.16819/j.1001-7216.2020.9056
2019-05-15;
2019-08-01。
国家自然科学基金资助项目(31701408);国家水稻产业体系资助项目(CARS-01-47);中国农业科学院农业科技创新工程资助项目(CAAS- ZDRW202011);中央级公益性科研院所基本科研业务费专项(Y2019PT19-01)。
