RNA-seq analysis identified hormone-related genes associated with prognosis of triple negative breast cancer
2020-05-25FeiChenYuanchengLiNaQinFengliangWangJiangboDuChengWangFangzhiDuTaoJiangYueJiangJunchengDaiZhibinHuChengLuHongbingShen
Fei Chen, Yuancheng Li, Na Qin, Fengliang Wang, Jiangbo Du, Cheng Wang,4,Fangzhi Du, Tao Jiang, Yue Jiang, Juncheng Dai, Zhibin Hu, Cheng Lu,✉, Hongbing Shen,✉
1Department of Breast Surgery, Women's Hospital of Nanjing Medical University, Nanjing Maternity and Child Health Care Hospital, Nanjing, Jiangsu 210004, China;
2Department of Epidemiology, Center for Global Health, School of Public Health, 3Jiangsu Key Lab of Cancer Biomarkers, Prevention and Treatment, Collaborative Innovation Center for Cancer Medicine, 4Department of Bioinformatics, School of Biomedical Engineering and Informatics, Nanjing Medical University, Nanjing, Jiangsu 211116, China;
5Department of Clinical Management, National Center for STD Control, Institute of Dermatology, Chinese Academy of Medical Sciences, Nanjing, Jiangsu 210042, China.
Abstract Triple negative breast cancer (TNBC) is an aggressive subtype of breast cancer that currently lacks effective biomarkers and therapeutic targets required to investigate the diagnosis and treatment of TNBC. Here we performed a comprehensive differential analysis of 165 TNBC samples by integrating RNA-seq data of breast tumor tissues and adjacent normal tissues from both our cohort and The Cancer Genome Atlas (TCGA). Pathway enrichment analysis was conducted to evaluate the biological function of TNBC-specific expressed genes. Further multivariate Cox proportional hazard regression was performed to evaluate the effect of these genes on TNBC prognosis. In this report, we identified a total of 148 TNBC-specific expressed genes that were primarily enriched in mammary gland morphogenesis and hormone levels related pathways, suggesting that mammary gland morphogenesis might play a unique role in TNBC patients differing from other breast cancer types. Further survival analysis revealed that nine genes (FSIP1, ADCY5, FSD1, HMSD, CMTM5, AFF3, CYP2A7, ATP1A2,and C11orf86) were significantly associated with the prognosis of TNBC patients, while three of them (ADCY5,CYP2A7, and ATP1A2) were involved in the hormone-related pathways. These findings indicated the vital role of the hormone-related genes in TNBC tumorigenesis and may provide some independent prognostic markers as well as novel therapeutic targets for TNBC.
Keywords: RNA-seq, triple negative breast cancer, prognosis, differential expression
Introduction
Breast cancer is the most diagnosed cancer and the leading cause of cancer-related death among females in the world, accounting for 25% of all cancer cases and 15% of all cancer deaths in women[1]. The incidence and mortality in China grew steadily in the last two decades[2], and the newly diagnosed breast cancer patients in China account for 12.2% of all newly diagnosed breast cancers worldwide, with 9.6%of deaths occurring in China[3]. Breast cancer is a heterogeneous disease with a variety of diversity in histologic, molecular and biological features[4],affecting the diagnosis and prognosis of breast cancer.Multiple molecular mechanisms involved in the course of it[5]. Triple negative breast cancer (TNBC) is a special subgroup characterized by lack of estrogen receptor (ER), progesterone receptor (PR) and human epidermal growth factor receptor 2 (HER2)expression. As patients diagnosed with TNBC cannot respond to established anti-hormone and anti-HER2 therapies[6–7], chemotherapy remains the only standard therapeutic approach. Although some new chemical regimens and target agents have been developed, the efficacy of these regimens in the treatment of TNBC remains unclear. Additionally, since TNBC holds many aggressive features compared with other subtypes of breast cancer, such as susceptibility in younger women, shorter period to relapse and higher tendency to metastasize to viscera rather than to bone[7–8], there is an urgent need to investigate potential biomarkers in the diagnosis and treatment of TNBC.
In addition to traditional clinicopathologic characteristics such as tumor size, the number of involved lymph nodes and fundamental biomarkers,microarray analysis has provided evidence that gene expression is important in the molecular classification and prognostic evaluation of breast cancer. Based on gene expression patterns, breast cancer can be classified into four main intrinsic subtypes including luminal-like, basal-like, HER2+and normal breast[9–10].And several gene expression signatures such as Mamma-Print[11]and Oncotype DX[12]have been developed to help predict the absolute benefit from a certain therapeutic regimen[13]. However, these signatures are valuable in patients with ER-positive breast cancer and less informative in ER-negative,high-proliferating breast tumors including TNBC[14].Molecular mechanisms underlying carcinogenesis of TNBC still need to be further explored to discover better prognostic markers and new therapeutic targets.In recent years, RNA sequencing has emerged as a new technology for high throughput transcriptome analysis[15]. Many studies have identified that differentially expressed transcripts, spliced genes, and splice isoforms can be potential biomarkers in the classification of breast cancer subtypes, which will be beneficial for the diagnosis and treatment of breast cancer[16–17].
In this study, we performed a comprehensive differential analysis of TNBC samples by integrating RNA-seq data from both our cohort and The Cancer Genome Atlas (TCGA). We included both paired TNBC and adjacent non-cancerous tissues as well as other breast cancer subtypes, aiming to detect a group of differentially expressed genes (DEGs) that could universally represent the molecular features of TNBC and be helpful in searching for prognostic and predictive markers of TNBC.
Materials and methods
Tumor samples collection and RNA extraction
Five paired breast tumors and matched adjacent normal tissues were obtained during surgical resection from the Maternity and Child Care Hospital of Nanjing Medical University. All tissues were snapfrozen. Informed consent was provided by all the subjects included in this study, which was approved by the Institutional Review Board of Nanjing Medical University. The tumor samples were microscopically confirmed to contain ≥70% invasive breast cancer cells by two independent pathologists, and the adjacent normal tissues contained no tumor cells.TNBC was confirmed according to the status of ER,PR, and HER2 based on immunohistochemical (IHC)classification. Briefly, tumors were considered negative for hormone receptors (ER and PR) if less than 1% of the tumor cells showed nuclear staining[18].HER2 status was evaluated based on the criteria from the HER2 testing guideline[19]. The negative status of HER2 was defined as HER2 IHC 0 or 1+; samples with HER2 IHC 2+ should be subsequently subjected toin situhybridization (ISH). Dako Envision immunohistochemistry staining system (Dako,Denmark) was employed for IHC testing. The primary antibodies were as follows: FLEX Monoclonal Rabbit Anti-Human Estrogen Receptor α (clone EP1, readyto-use, Dako), FLEX Monoclonal Rabbit Anti-Human Progesterone Receptor (clone PgR636, ready-to-use,Dako), anti-HER2/neu (4B5) Rabbit Monoclonal Primary Antibody (VENTANA, USA). PathVysion HER2 DNA Probe Kit (Vysion, USA) was used for ISH assays. All the collected tissues were preserved in RNAlater solution (Ambion, USA) and stored at –80°C prior to RNA extraction. The anamnestic and clinical-pathological characteristics of the 5 TNBC patients were shown inSupplementary Table 1(available online). Total RNA was extracted from frozen breast cancer tissue samples using the RNeasy Mini Kit (Qiagen, Germany) according to the manufacturer's instructions. RNA pellets were resuspended in nuclease-free water, and the concentration was quantified using the RNA BR Assay kit for the Qubit 2.0 fluorimeter (Life Technologies, UK).
cDNA synthesis, library construction, and RNAseq
RNA quality was evaluated using the Agilent 2100 Bioanalyzer system (Agilent Technologies, USA).Only high-quality RNAs (RIN≥7.5) were selected for cDNA library construction subsequently. The cDNA libraries were prepared using the TruSeq RNA sample prep kit (Illumina, USA) and subsequently amplified in flowcells using Illumina cBot. Finally, standard Illumina RNA-seq protocol according to the manufacturer was conducted to generate the transcription profiles of the samples using the Illumina HiSeq 1500 (Illumina) for 2×101 cycles.
Transcriptome assembly, gene annotation and mapping
The FastQC version 0.11.8 was used to access the quality score distribution of the sequencing reads, and the low-quality reads (Phred score ≤20) were removed using Trimmomatic V0.32 prior to the analysis. The remaining qualified reads were aligned to the GENCODE Version 19 genome assembly using the TopHat (version 2.1.0) and BOWTIE (version 2.3.4.3), allowing two mismatches in the alignment for each read. Differential expression analysis of TNBC was performed with R 3.1.1 Bioconductor package DESeq2. And the batch effects were adjusted by Bioconductor package sva. URLs of the software mentioned above were presented inSupplementary Table 2(available online).
Preparation of TCGA expression and clinical data
We obtained the expression data and clinical information of breast cancers from TCGA Firehose Broad GDAC (http://gdac.broadinstitute.org/, version 2016-01-28 release). The breast cancer types, TNBC,ER+, and HER2+, were classified based on the IHC results included in the clinical data. Briefly, ER+were defined as samples with ER positive, HER2 negative,and PR positive or negative, HER2+samples were those with HER2 positive, ER and PR positive or negative, and TNBC samples were negative for all three receptors (ER/PR/HER2 all negative). In all, we included 595 ER+breast cancer samples, 185 HER2+breast cancer samples and 160 TNBC samples(including 14 samples with paired adjacent noncancerous tissues available) in the following analysis.Clinical information on the cases obtained from the TCGA database was presented inSupplementary Table 3(available online).
Differential gene expression analysis and pathway enrichment analysis
Differential expression analysis of TNBC was performed with R 3.1.1 Bioconductor package DESeq2. The following workflow was carried out for the comparison of TNBC tumor tissues versus nontumor tissues or other two breast cancer types. We first removed genes with normalized read counts <5 in less than 20% samples. Raw read counts were normalized by DESeq2. The magnitude (log2transformed fold change) and significance (P-value)of differential expression between groups were calculated, and genes with false discovery rate (FDR)adjustedP-values <0.05 were considered as significant. We obtained DEGs with the following criteria: FDR<0.05 and |log2fold change|≥1. Genes significantly differentially expressed between TNBC tumor samples and both adjacent normal samples and other two breast cancer types were defined as TNBCspecific expressed genes.
A pathway enrichment analysis was performed using the R 3.1.1 Bioconductor package clusterProfiler for the candidate TNBC-specific differential expressed genes. For multiple test correction,P-values were adjusted by the Benjamini-Hochberg false discovery rate (BH-FDR). The workflow was shown inFig. 1.
Statistical analysis
Kaplan-Meier method and multivariate Cox proportional hazard regression analyses were performed to evaluate the association between expression of TNBC-specific expressed genes and breast cancer prognosis, with adjustment of age and clinical stage. Patients were divided into two groups:patients with gene expression above the median were defined as "high expression group", which represented a group of patients expressed relative high gene expression, and patients with gene expression below median level were defined as "low expression group".The crude or adjusted hazard ratio (HR) and their 95%confidence intervals (95% CI) were calculated. All tests were two-sided and all the significance level was set at FDR<0.05. We used the p. adjust function(based on BH-FDR) in R software to adjustPvalues.All the statistical analyses were carried out with R 3.1.1 software (http://www.cran.r-project.org/).
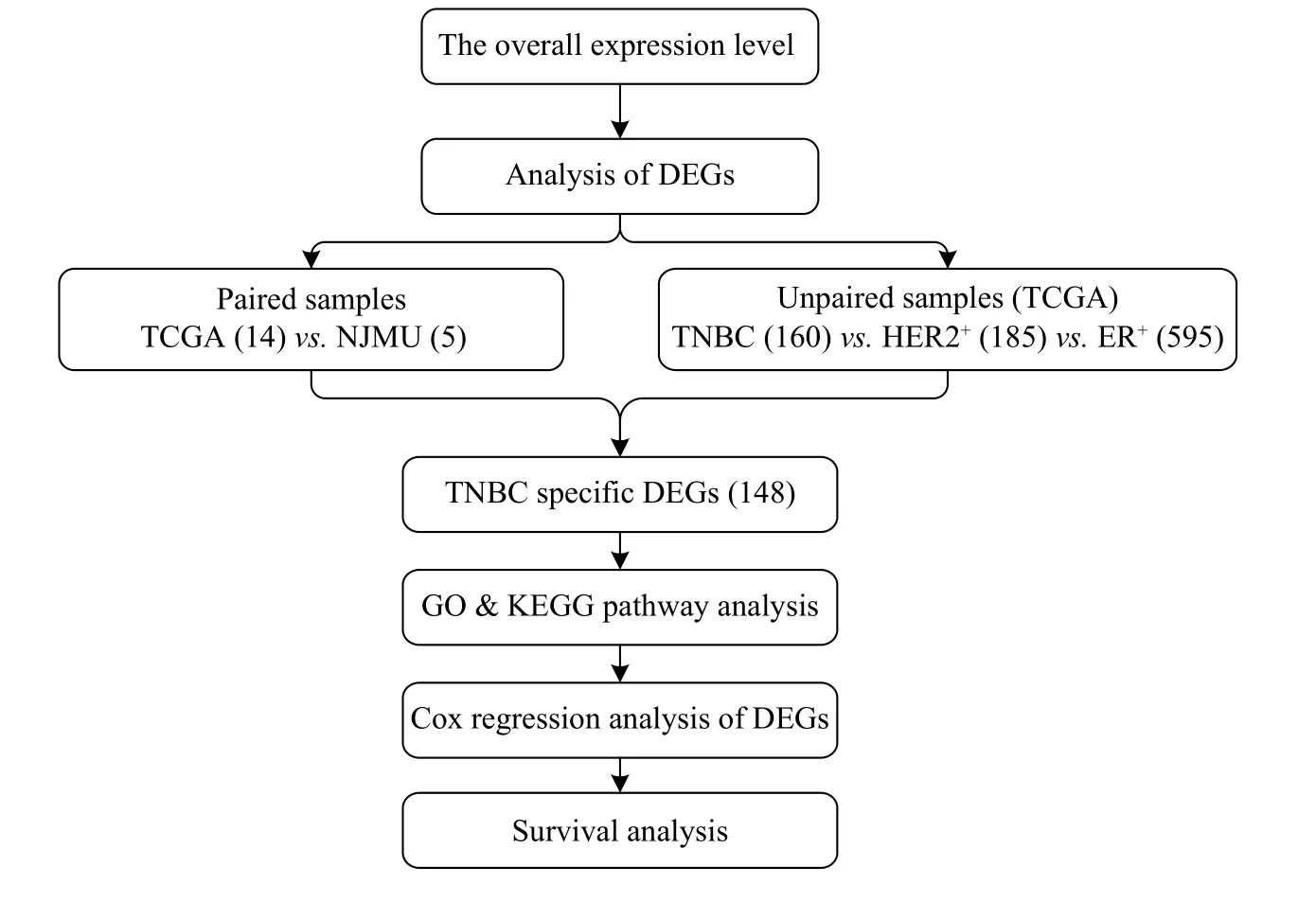
Fig. 1 Overview of the steps involved in the RNA-seq analysis of TNBC, HER2+, and ER+ breast cancers. DEGs: differentially expressed genes; TNBC: triple negative breast cancer; TCGA: The Cancer Genome Atlas; NJMU: Nanjing Medical University.
Results
Characterizations of sequencing and mapping
A total of 347.3 and 357.9 million reads were obtained from 10 independent libraries of paired breast cancer and adjacent non-cancerous tissues,respectively. Most reads reached Phred-like quality scores (Q-scores) at the Q30 level, indicating that the probability of an incorrect base call is 0.001%. The average coverage of sequencing depth reached approximately 53.45× of the human transcriptome.After alignment using TopHat and BOWTIE, 98.57%to 99.12% uniquely aligned reads of five paired tissues could be mapped to the human reference genome (Supplementary Table 4, available online).
Differential gene expression analysis
We first performed paired differential gene expression analysis, and 2 680 genes were found to be differentially expressed in 5 paired TNBC samples from our cohort, 1 977 genes were found in 14 paired TCGA samples and 894 genes were found in the same differential direction in the above two differentially expressed gene sets. Then, we further compared the transcriptome profiles between 160 TNBC samples and 595 ER+and 185 HER2+breast cancer samples respectively. We identified 741 and 1 074 DEGs between TNBC and the other two subtypes respectively. By integrating the tumor-adjacent differential expressed genes defined above, we identified a total of 148 TNBC-specific expressed genes [Fig. 2,Supplementary Fig. 1andSupplementary Table 5(available online)].
Gene Ontology and KEGG enrichment analysis
To evaluate the potential functions of the 148 TNBC-specific expressed genes defined in our study,we further performed enrichment analysis with the Kyoto Encyclopedia of Genes and Genomes (KEGG)pathways and Gene Ontology (GO) terms. We identified that these genes were significantly enriched in 5 KEGG pathways and 33 GO terms. Interestingly,most of the enriched pathways and GO terms were related to the mammary gland morphogenesis and hormone levels related pathways, such as mammary gland duct morphogenesis, mammary gland epithelium development, steroid metabolic process and its like, suggesting that mammary gland morphogenesis might play a unique role in TNBC patients compared from other breast cancer types(Table 1and2).
Survival analysis
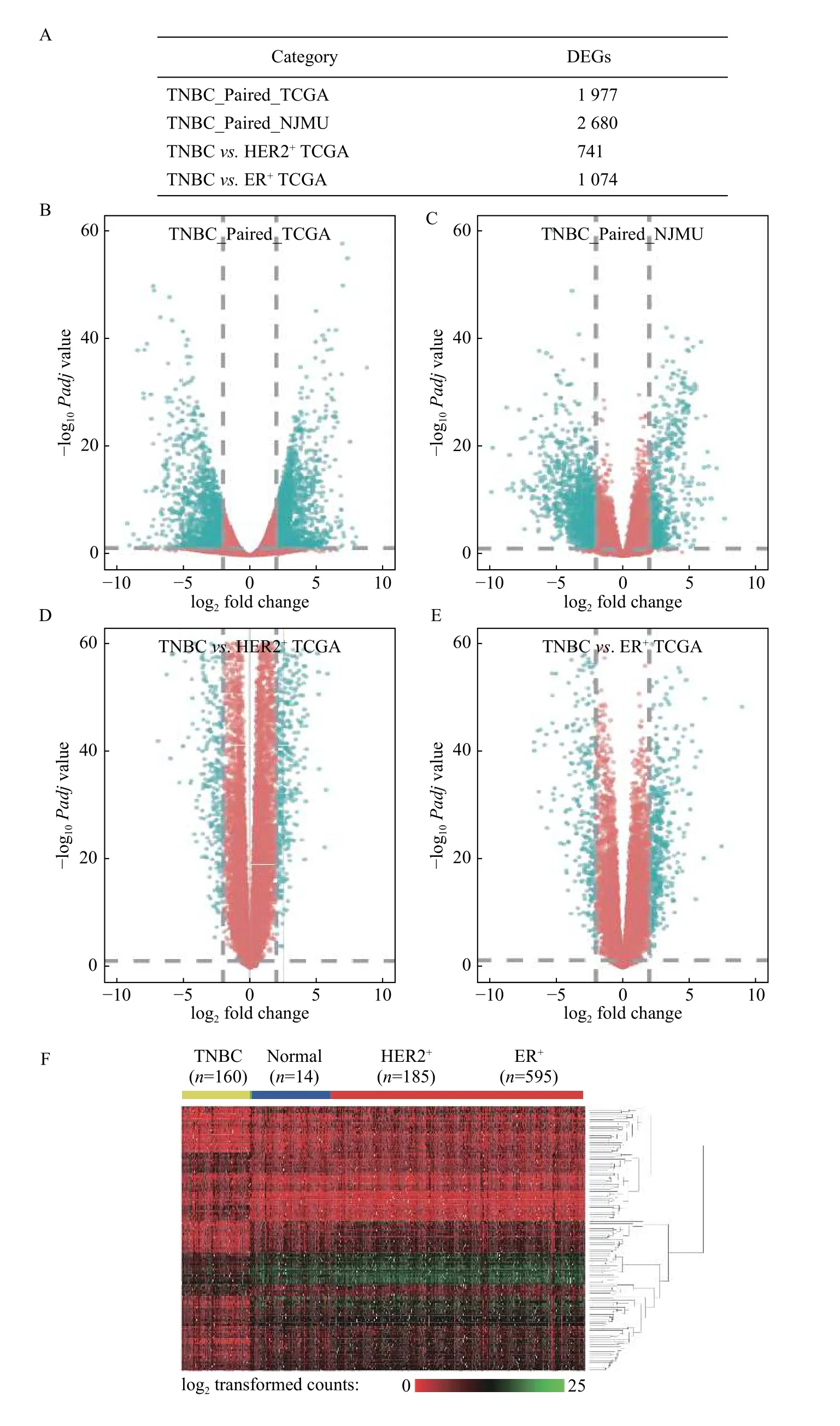
Fig. 2 Differentially expressed genes between TNBC, HER2+, and ER+ breast cancers. A: The number of DEGs (FDR<0.05 and |log2 fold change|≥1) among TNBC paired in TGCA, TNBC paired in NJMU, TNBC vs. HER2+ in TCGA, and TNBC vs. ER+ in TCGA. B–E:Volcano plots for DEGs in TNBC paired in TGCA (B), TNBC paired in NJMU (C), TNBC vs. HER2+ in TCGA (D), and TNBC vs. ER+ in TCGA (E). Red: genes with FDR<0.05 and |log2 fold change|≥1; Blue: genes with FDR≥0.05 or |log2 fold change|<1. F: Heatmap of 148 TNBC-specific expressed genes in TCGA TNBC (n=160), adjacent normal tissues (n=14), HER2+ (n=185), and ER+ samples (n=595).Green: Relative low log2 transformed counts; Red: Relative high log2 transformed counts. DEGs: differentially expressed genes; TNBC:triple negative breast cancer; TCGA: The Cancer Genome Atlas; NJMU: Nanjing Medical University.

Table 1 Top 10 KEGG pathways revealed in TNBC-specific expressed genes
Then, we further evaluate the prognostic effect of 148 TNBC-specific expressed genes in 145 TNBC cases from TCGA with survival information available.Kaplan-Meier method and multivariate Cox proportional hazard regression were performed. The base-line and follow-up information of the 145 patients was shown inSupplementary Table 6andSupplementary Fig. 2(available online). Interestingly,nine genes were found to be significantly associated with prognosis of TNBC patients, includingFSIP1(HR=4.61,P=6.92E-03),ADCY5(HR=0.22,P=7.53E-03),FSD1(HR=0.20,P=8.11E-03),HMSD(HR=0.24,P=1.13E-02),CMTM5(HR=0.28,P=1.94E-02),AFF3(HR=0.29,P=2.63E-02),CYP2A7(HR=2.99,P=3.36E-02),ATP1A2(HR=2.86,P=4.73E-02), andC11orf86(HR=0.34,P=4.98E-02). Three of these nine genes were previously reported to be involved in the hormonerelated pathways, includingADCY5,CYP2A7, andATP1A2[Table 3,Fig. 3, andSupplementary Fig. 3(available online)].
Discussion
TNBC accounts for approximately 15% of all invasive breast cancers and is recognized as an aggressive subgroup as it occurs primarily in young women and has a high potential to metastasize[7].Currently, chemotherapy remains the mainstay of treatment for patients with TNBC for the absence of definite therapeutic targets, so it is of an urgent need to identify potential diagnostic and therapeutic markers such as immunotherapy[20]. Based on microarray and deep sequencing techniques,molecular alterations in TNBC have been widely explored including deficiency in homologous recombination, increased immunological infiltration,and expression of androgen receptors, leading to great progress in the clinical trials. However, the new drugs have been failed to be validated in phase 3 trials[21],leaving TNBC the most complex subset of breast carcinoma.
In the present study, we conducted a comprehensive differential expression analysis by comparing the expression profiles of TNBC samples with adjacent normal samples, ER+breast cancers, and HER2+breast cancers respectively. We identified a total of 148 TNBC-specific expressed genes, GO term enrichment analysis and KEGG pathway analysis found these genes were primarily enriched in mammary gland morphogenesis and hormone levels related pathways. Further survival analysis revealed that nine genes were significantly associated with the prognosis of TNBC patients.
GO term enrichment analysis suggested that most of the TNBC-specific expressed genes were enriched in steroid metabolic process and regulation of hormone levels. It has been well known that steroids induce both chronic and acute actions within cells, and mounting evidence shows that sex steroids are involved in cell growth and proliferation[22–23]. In someprior studies, steroid mediators (i.e.serum estrogens and androgens) were found to be associated with breast cancer risk[24]. Of note, estrogens might accelerate the development of breast cancer at many points along with the progression from early mutation to tumor metastasis. By increasing cell proliferation,estrogens may also decrease the capability of DNA damage repair[25]. Furthermore, estrogens might have genotoxic effectsviatheir reactive metabolites[25].Additionally, among TNBC patients, some investigators have reported that steroid Receptor Coactivator-3 performs as a therapeutic target due to its regulation of cellular signaling pathways that are vital for cancer proliferation and development, which can support our results as well[26]. Consequently, we can initially conclude that it is biologically plausible for us to observe the differential genes mainly involved in the steroid metabolic process in TNBC patients.
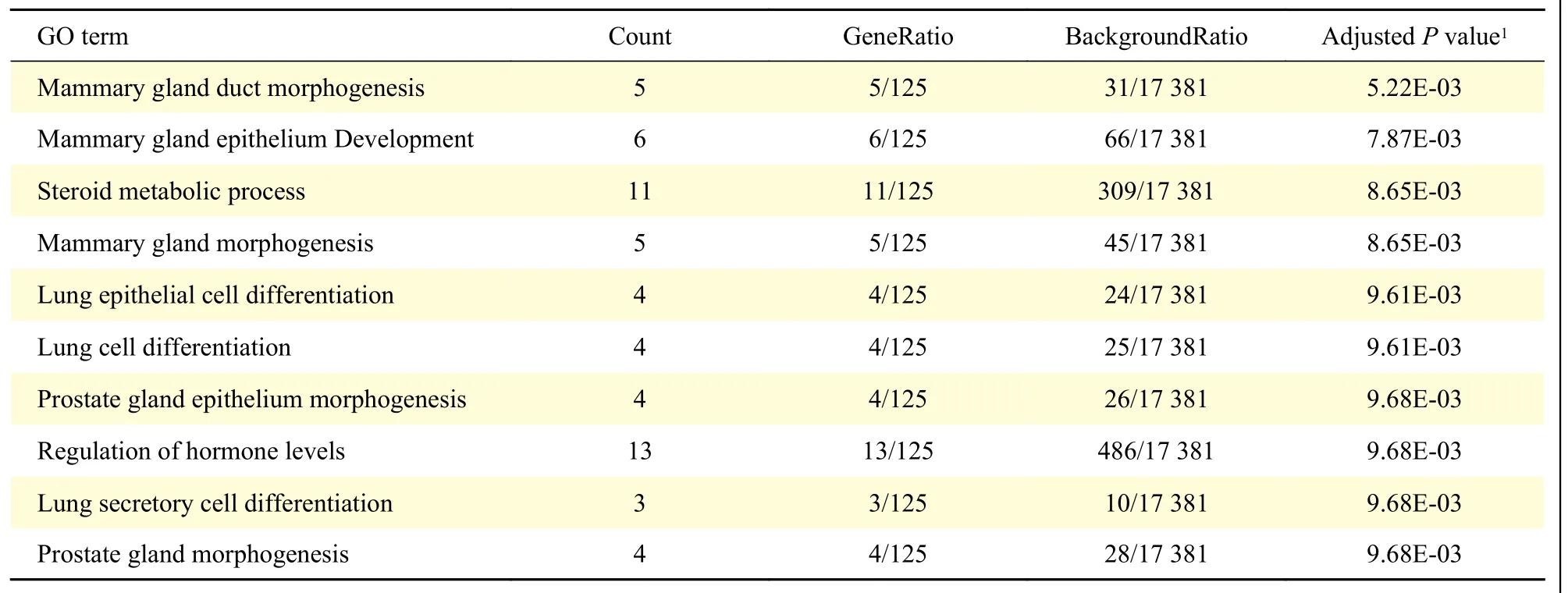
Table 2 Top 10 GO terms revealed in TNBC-specific expressed genes

Table 3 Survival analysis of nine DEGs that significantly associated with prognosis of TNBC patients in TNBC, HER2+, and ER+patients
Among those genes,FSIP1, testicular and spermatogenesis-related antigen is expressed in abundance in various cancers, including breast cancer,lung cancer,etc. The level ofFSIP1is low or undetectable in most normal tissues except the testis[27]. In our study, we observed the prognostic value ofFSIP1in TNBC. High expression ofFSIP1was associated with a poorer outcome in TNBC, and reversely, a better outcome in ER+patients. Consistent with our findings, several lines of evidence have previously indicated thatFSIP1expression in breast cancer correlated negatively with survival[28]. Indeed,FSIP1-mediated alterations in microtubule and dynein function may support the microtubule network and enhance mitotic robustness in cancer cells.Additionally, it is reported thatFSIP1can bind to HER2 directly in HER2+breast cancer to promote cancer progression[27]. However, we didn't find a significant association betweenFSIP1and prognosis in HER2+breast cancer, which may due to a limited number of samples. As the correlation between ER status andFSIP1was inconsistent among papers,larger sample studies andin vivoassays are needed to further validate[28–29].
CMTM5, a member of the CMTM protein family, is located at 14q11.2 and has been reported to act as a tumor-suppressor gene since it was specifically downregulated in numerous human cancers[30].Previous studies also revealed that CMTM family proteins, including CMTM7, could inhibit cell growth and induce apoptosis[31]. Xuet al[30]reported that low expression ofCMTM5in hepatocellular carcinoma significantly correlated with poor overall survival.Interestingly, our study also suggested the prognostic value ofCMTM5and it might prolong the survival of patients with TNBC.
AFF3, a lymphoid-specific gene, encodes a 1227-amino acid protein that is presumed to play an important role in transcriptional regulation[32]. In our study, expression ofAFF3was observed to be associated with improved survival among patients with TNBC. Inconsistent with our results,AFF3was showed to be related to worse overall survival in ER+breast cancer treated with tamoxifen in previous studies[33]. No data to date is available regarding the role ofAFF3in TNBC. Hence, further investigation is required in terms of the biological mechanism and the prognostic value ofAFF3in TNBC.
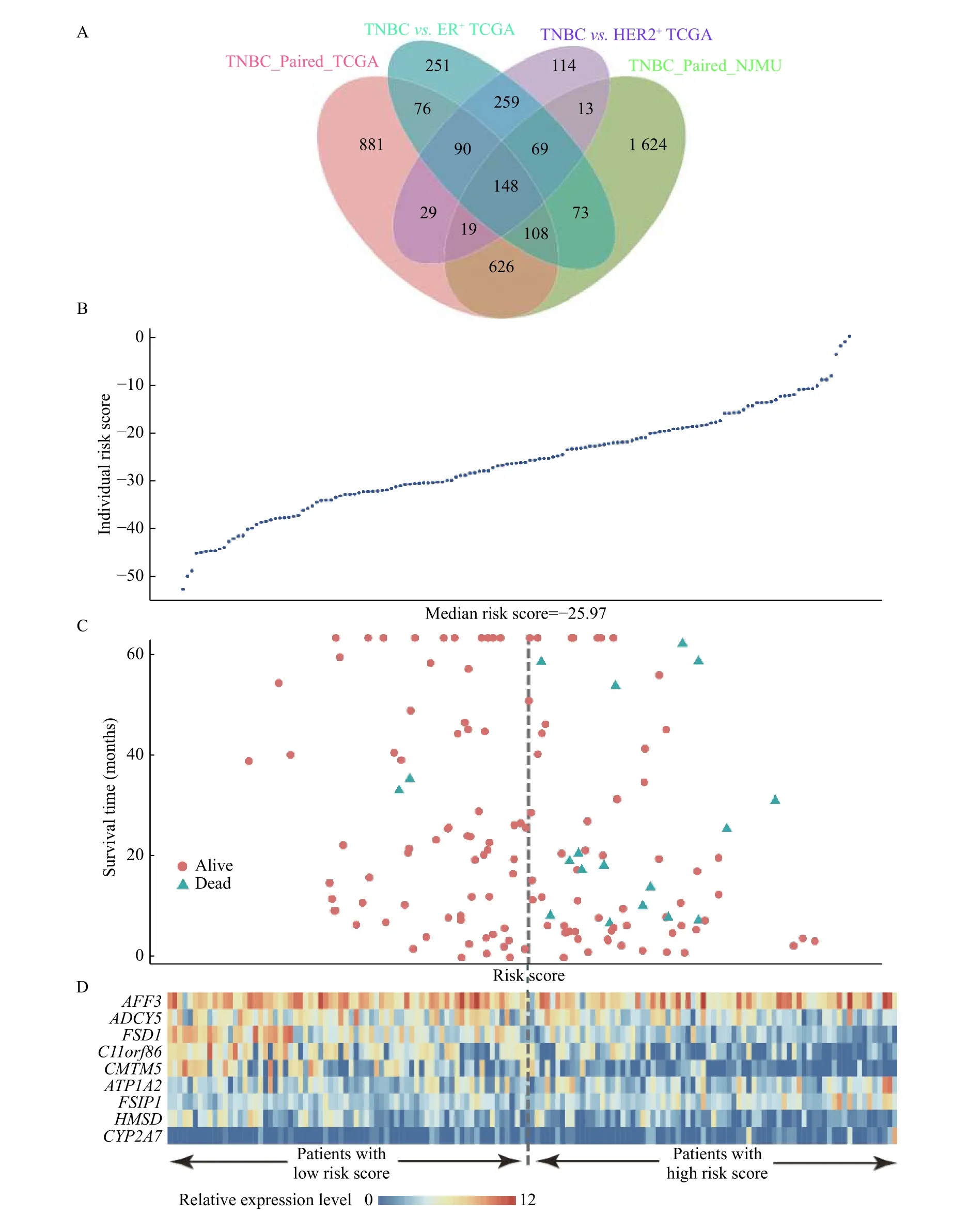
Fig. 3 Differential gene expression analysis. A: Veen plot of 148 TNBC-specific overlapped expressed genes in TNBC paired in NJMU,TNBC paired in TCGA, TNBC vs. HER2+ in TCGA, and TNBC vs. ER+ in TCGA. B: Individual risk scores of 145 TNBC patients with survival information in TCGA. C: Survival time and risk scores of 145 TNBC patients with survival information in TCGA. D: The expression of nine survival-related genes among 145 TNBC patients with survival information in TCGA.
ATP1A2encodes the α2 subunit of the Na+/K+-ATPase (NKA) that is abundantly expressed in skeletal muscle and brain astrocytes. Some studies have demonstrated the anticancer effect of cardiac glycosides that directly inhibit NKA activity[34].Down-regulation ofATP1A2expression was previously reported in breast cancer[35]. In our study,we observed overexpression ofATP1A2was associated with favorable outcome in TNBC. Given that the prognostic role ofATP1A2in breast cancer remains unclear, more researches are required with respect to the effect ofATP1A2on the occurrence and progression of breast cancer.
In addition, we observed that geneADCY5, FSD1,HMSD, C11orf86are down-regulated in TNBC with prolonging survival, whereas geneCYP2A7is associated with poorer survival. To our knowledge,ADCY5encodes adenylate cyclase 5, which catalyzes the generation of cAMP in type 2 diabetes[36].FSD1encodes a centrosome associated protein that is characterized by an N-terminal coiled-coil region downstream of B-box (BBC) domain, some studies found that the methylation ofFSD1may play a role in Epstein-Barr virus-associated gastric carcinomas[37].CYP2A7encodes a member of the cytochrome P450 superfamily, and cytochrome P450 metabolites are proved to play a critical role in carcinogenesis[38]. No data to date are available regarding the association between these genes and breast cancer. Thus, the results of our study are warranted to be further confirmed.
In conclusion, we identified a group of TNBCspecific expressed genes by comparing gene expression of TNBC tumors with that of paired normal tissues and non-TNBC tumors. Most of these genes were involved in the hormone-related pathways.Survival analysis further confirmed that nine steroid hormone-related genes were independent prognostic markers in TNBC. These findings emphasized the vital role of the hormone-related genes in TNBC tumorigenesis and may provide new therapeutic targets for TNBC.
Acknowledgments
We would like to thank all the people participated in this study. This work was supported by the Nanjing Medical Science and Technique Development Foundation (ZKX17041), the Natural Science Foundation of Jiangsu Province (BK20161120), and the Maternal and child health research project of Jiangsu Province (F201628), the Priority Academic Program Development of Jiangsu Higher Education Institutions (Public Health and Preventive Medicine)and Top-notch Academic Programs Project of Jiangsu Higher Education Institutions (PPZY2015A067).
杂志排行

THE JOURNAL OF BIOMEDICAL RESEARCH的其它文章
- Angiosarcomatous component in gliosarcoma: case report and consideration of diagnostic challenge and hemorrhagic propensity
- Daily high-dose aspirin does not lower APRI in the Aspirin-Myocardial Infarction Study
- The level of bile salt-stimulated lipase in the milk of Chinese women and its association with maternal BMI
- AAV-mediated human CNGB3 restores cone function in an allcone mouse model of CNGB3 achromatopsia
- Inhibitory role of peroxiredoxin 2 in LRRK2 kinase activity induced cellular pathogenesis
- H2S protects against diabetes-accelerated atherosclerosis by preventing the activation of NLRP3 inflammasome