Inhibitory role of peroxiredoxin 2 in LRRK2 kinase activity induced cellular pathogenesis
2020-05-25KangYanWenfengZhangXuHanFeiChangYongjianLiu
Kang Yan, Wenfeng Zhang, Xu Han, Fei Chang,✉, Yongjian Liu,2,✉
1Jiangsu Key Laboratory of Xenotransplantation, School of Basic Medical Science, Nanjing Medical University,Nanjing, Jiangsu 211166, China;
2Department of Pharmacology & Chemical Biology, University of Pittsburgh School of Medicine, Pittsburgh, PA 15216, USA.
Abstract
Keywords: peroxiredoxin 2, LRRK2, Parkinson's disease
Introduction
Parkinson's disease (PD) is the second most common neurodegenerative disease. One of the wellknown genetic causes of PD is leucine-rich repeat kinase 2 (LRRK2), which is a multi-domain protein containing a Ras of complex proteins (Roc)-GTPase domain, a C-terminal of Roc (COR) domain, and a MAPKKK-like kinase domain[1]. It has been widely accepted that mutations within these functional domains, such as R1441C mutation in the Roc domain and G2019S in the kinase domain[2–3], are closely associated with both familiar and sporadic PD.However, the molecular and cellular mechanisms underlying the pathogenesis of LRRK2-mediated PD are still elusive. As the most common PD-associated LRRK2 mutation, G2019S has been reported to have elevated kinase activity which may promote toxicity in neuronal cells[4–5]. Another R1441C mutation with increased GTP binding to Roc-GTPase also enhances 2019, Epub 31 July 2019
CLC number: R742.5, Document code: A
The authors reported no conflict of interests.
This is an open access article under the Creative Commons Attribution (CC BY 4.0) license, which permits others to distribute, remix,adapt and build upon this work, for commercial use, provided the original work is properly cited.the kinase activity through a potential intracellular regulatory mechanism[4,6], indicating that elevated kinase activity might be the major contributor to the LRRK2-mediated pathogenesis of PD. Nonetheless,how the LRRK2 kinase activity and its toxicity in PD are regulated remains largely unclear.
Previous studies have reported that several interactors of LRRK2 are involved in the important cellular processes, including vesicular trafficking,cytoskeletal function, and autophagy, and sequentially participate in the pathogenesis of PD[7]. For example, a subset of Rab GTPases, the master regulators of membrane trafficking, has been identified as LRRK2 substrates membrane trafficking[8]. On the other hand,as the interactor of LRRK2, Rab7L1/Rab29 has been shown to be involved in LRRK2 mediated membrane trafficking and neurite outgrowth[9]. Interestingly, in addition to being an LRRK2 substrate, Rab29 may also act as an upstream regulator to activate LRRK2 kinase activity on downstream effectors like Rab8 and Rab10 by recruiting LRRK2 to the membrane of the Golgi complex[10]. Moreover, LRRK2 mutations have been shown to cause neurite branching abnormality,and further studies have suggested that such morphological change may be consequences of LRRK2-modulation of cytoskeletal dynamics by associating with tubulin and actin[11]. Thus, the identification of novel interacting protein has provided tremendous insight into the understanding function and its regulation of LRRK2 in PD pathogenesis[12].
Recently we performed a yeast-two hybrid (Y2H)screening using Roc-COR domain as bait and identified a novel interactor peroxiredoxin 2 (Prx2), a member of the Prx family of antioxidant enzymes.Since it has been well documented that oxidative stress contributes to the pathogenesis of PDviaelevating the reactive oxygen species (ROS) activity to induce mitochondrial damage in DA neurons[13–14],the novel interactor Prx2 as part of ROS scavenger family proteins seems well suitable to the signaling pathway in LRRK2 toxicity[15]. More importantly,animal-based studies suggested that overexpression of Prx2 exhibited neuroprotection against 6-OHDA toxicity in DA neurons and also showed anti-apoptotic effectsviasuppression of ASK1-dependent activation of the JNK/c-Jun and p38 pro-death pathways[16].Interestingly, another member of this family Prx3 was also reported to interact with LRRK2[17], supporting the notion that oxidative stress and related molecular mechanisms may be part of LRRK2 induced cellular toxicity.
In this study, we have shown the neuronal specific distribution of Prx2 with preferential expression in dopaminergic neurons using both RT-PCR and immunostaining approaches. We also demonstrated that Prx2 interacts specifically with the COR domain of LRRK2 and dramatically reduces its kinase activity[12]. Furthermore, overexpressed Prx2 could rescue the transfected cell from LRRK2G2019Smutant induced apoptosis and reverse the altered retrograde membrane trafficking.
Materials and methods
Reagents and antibodies
Cycloheximide (CHX) and poly-D-lysine were purchased from Sigma Chemical Co. (USA). Matrigel was purchased from Becton Dickinson (USA). Other reagents were purchased from Thermo Fisher (USA):Protein A/G Agarose, Dulbecco's Modified Eagle Medium (DMEM), Lipofectamine 2000,penicillin/streptomycin and TRIzol. Cosmic calf serum (CCS) and fetal bovine serum (FBS) were from HyClone (USA). All the restriction enzymes and products related to PCR were from NEB (USA). The following primary antibodies were used: mouse monoclonal anti-HA and rabbit polyclonal anti-HA(Covance, USA), mouse monoclonal anti-Flag(Sigma, USA), mouse monoclonal anti-Myc (Santa Cruz Biotechnology, USA), rabbit polyclonal anti-Rab10 pT73 and mouse monoclonal anti-LC3 A/B(Abcam, UK). All secondary antibodies used in this study were purchased from Thermo Fisher (USA):Alexa 488-conjugated donkey anti-mouse secondary antibody, Alexa 488-conjugated donkey anti-rabbit secondary antibody, Alexa 568-conjugated donkey anti-mouse secondary antibody, Alexa 568-conjugated donkey anti-rabbit secondary antibody, Alexa 647-conjugated donkey anti-rat secondary antibody, HRPconjugated goat anti-mouse secondary antibody, and HRP-conjugated goat anti-rabbit. siRNA of Prx2 was designed and produced by Shanghai Genechem Co.,Ltd. (China).
Cell culture and transfection
There are 3 cell lines used in the experiments: COS-7, HeLa, and SH-SY5Y cells. COS-7 cells and HeLa cells were cultured in DMEM supplemented with 10%CCS and 1% penicillin/streptomycin. SH-SY5Y cells were cultured in DMEM supplemented with 10% FBS and 1% penicillin/streptomycin. All cells were maintained in the incubator at 37 °C with 5% CO2.
DNA constructs
The pcDNA3.1-3HA-LRRK2 and its variants were described previously[9]. The cDNA fragments in the plasmids of pcDNA3.1-3HA-Roc, pcDNA3.1-3HACOR and pcDNA3.1-3HA-ROCCOR were generated by proof-reading PCR and then inserted into corresponding vectors. Human cDNAs for Prx2 and 3 were obtained from Open Biosystem (Dharmacon,Lafayette, USA) and inserted into inhouse pcDNA3.1-Myc plasmid.
Animals
The use of animals was approved by the Institutional Animal Care and Use Committee of Nanjing Medical University. The animals were housed in stainless steel cages with wood bedding to minimize additional exposure to endocrine disrupting chemicals [(temperature of (23±2) °C, humidity of(55±5)%, 12:12 hours light/dark cycle, and lights from 06:00 a.m.] in Animal Research Center of Nanjing University. They had free access to food and water.Adult Sprague-Dawley rats (male) were used in this study. Brain regions [cortex, hippocampus, and substantia nigra (SN)] were isolated according to the 3rdedition of The Rat Brain in Stereotaxic Coordinates.
Reverse transcription polymerase chain reaction(RT-PCR)
Total RNA of cells or brain tissues was extracted with Trizol following the manufacturer's protocol. A total of 1 μg RNA was reversely transcribed to cDNA using a Prime Script RT reagent kit (Vazyme Biotech,China). PCR was performed with primers listed inSupplementary Table 1, available online. PCR products were separated by 1% agarose gel.
Co-immunoprecipitation and protein half-life analysis
Cells transiently transfected with respective plasmids were lysed in lysis buffer (0.5% NP-40, 150 mmol/L NaCl, 50 mmol/L Tris-HCl, pH 7.0 and 5 mmol/L EDTA in PBS) supplemented with 1 mmol/L PMSF and 0.1 mmol/L leupeptin for 10 minutes on ice. Cell lysate was centrifuged at 1 600gand the supernatants were then precleared by incubation for 60 minutes at 4 °C with 30 μL protein A/G agarose beads and centrifugation at 8 000gfor 5 minutes. The precleared lysates were incubated for 2 hours at 4°C with 30 μL protein A/G agarose beads bound to polyclonal antibody to tagged protein. After immunoprecipitation, the beads were washed 4 times with wash buffer (0.5% NP-40, 150 mmol/L NaCl, 50 mmol/L Tris-HCl, pH 7.0 and 5 mmol/L EDTA) and then processed for Western blotting assay.
For half-life detection, HeLa cells were transiently transfected with respective plasmids. After 24 hours,cells were treated with 100 μg/mL CHX which was used to block protein synthesis and samples were collected at the time interval of 0, 6, or 12 hours.Protein expression levels were analyzed by Western blotting assay.
Western blotting analysis
Proteins were separated by 10% SDS-PAGE before electrotransfering to nitrocellulose (Millipore, USA).The filters were then blocked in PBS containing 0.1%Tween-20 (TBS) and 5% nonfat dry milk, incubated in TBS with 1% nonfat dry milk and primary antibody at dilutions from 1:1 000 to 1:10 000 for 60 minutes at room temperature, washed in TBS, and incubated in appropriate secondary antibody conjugated to peroxidase for additional 60 minutes followed by washing in TBS and visualization by enhanced chemiluminescence with the Tanon 5200 gel imaging system (Tanon, China).
Immunofluorescent staining, TUNEL and confocal microscopy
Immunofluorescent staining was performed as previously described[18]. Briefly, cells were plated onto glass coverslips coated with poly-D-lysine and Matrigel before transfection using Lipofectamine 2000 according to the manufacturer's instructions.Twenty-four hours later, cells were fixed with 4%paraformaldehyde followed by blocking in blocking buffer (2% BSA, 1% fish skin gelatin and 0.02%saponin in PBS). Cells were then incubated with primary antibody in blocking buffer for one hour at room temperature and further incubated with the appropriate secondary antibody for one hour.
Detection of apoptotic cells was performed using TUNEL Apoptosis Detection Kit (Vazyme, China)according to the manufacturer's instructions. All images were visualized with a confocal laser microscope (LSM710, Zeiss) and the images processed using the NIH Image program and ZEN program.
Statistical analysis
Statistical analysis was performed using the GraphPad Prism software (version 5.0, GraphPad Software). For quantitative neurite length analysis was determined by unpairedt-test as indicated, and the expression levels of endogenous in half-life studies were quantified by densitometry of the bands between two treatment groups, and statistical significance was performed by one-way ANOVA followed by Tukey's post hoc test, denoting * ifP<0.05, ** ifP<0.01 and*** ifP<0.001. Results are expressed as mean±SEM if not indicated otherwise.
Results
Preferential distribution of Prx2 in brain regions and neuronal cells
Previous studies have shown that Prx2 is a key modulator in neuroprotection by clearing ROS[19]. To confirm whether Prx2 is specifically expressed in neuronal cells, we compared Prx2 expression level in non-neuronal cell line MDA-MB-231 and neuronal SH-SY5Y cells by RT-PCR. As shown inFig. 1AandB, Prx2 expression level in SH-SY5Y cells was higher than that in MDA-MB-231 cells. Interestingly, our result also suggested that in SH-SY5Y cell line, Prx2 showed a higher expressing level than the neuronal cell-related Prx3[17].
To investigate the Prx2 expression in different brain regions, we isolated the cortex, hippocampus and SN from the brain of SD rats, and examined the mRNA levels of all the members (Prx 1-6) of peroxiredoxin family in these tissues by RT-PCR. GAPDH was used as semi-quantitative internal reference while tyrosine hydroxylase (TH) was the indicator of dopaminergic neurons. As shown inFig. 1CandD, Prx2 was not only highly expressed, but also showed higher expression levels than Prx3 in all the three regions,especially in SN of midbrain tissue. Furthermore, to confirm whether Prx2 protein is highly expressed in TH-positive neuronal cells, we used antibodies to costain TH cells and endogenous Prx2 in SD rat brain slices and found that Prx2 was preferentially expressed in TH positive cells (Fig. 1E). Taken together, these data suggested that Prx2 was preferentially expressed in neuronal cells.
Prx2 specifically interacts with LRRK2
Our Y2H screening suggested that Prx2 might be a novel interactor of LRRK2 (data not shown). To confirm this screening data, we performed coimmunoprecipitation (co-IP) assay to biochemically demonstrate that Prx2 could specifically bind with LRRK2 (Fig. 2A). Interestingly, it has been reported that Prx3 also interacted with LRRK2[17], which was also confirmed in our study by co-IP assay. However,we noticed that Prx3 showed a much weaker interaction with LRRK2 (Fig. 2A). Since LRRK2 contains several domains (Fig. 2B) and domain dependent interaction would imply their functional consequence, we next figure out which one mediated the interaction with Prx2. Based on our previous result of Y2H in which Prx2 might interact with Roc-COR domain of LRRK2, we made several truncated LRRK2 containing Roc, COR and Roc-COR domains to perform co-IP assay respectively. As shown inFig. 2C,Prx2 only interacted with Roc-COR and COR, but not Roc domain, strongly suggesting that LRRK2-COR domain is required and sufficient for the interaction with Prx2. The Roc-COR domain of LRRK2 belongs to the family of small G-proteins which are GTP binding proteins switching between an active GTPand inactive GDP-bound state[20]. Thus, this domain related binding may suggest that Prx2 participates in the Roc-GTPase mediated function.
LRRK2 mutations are the major cause of inherited and sporadic PD and the G2019S mutation within the kinase domain is the most common LRRK2 mutation,which increases the kinase activity two to three folds and thus subsequently causes substrates hyperphosphorylation. Previous studies have reported that Roc-COR domain is essential for kinase activity,implying a present cross-talk between kinase and GTPase domains intracellularly[21]. Thus, we hypothesized that regulator of the GTPase domain might influence the activity of the kinase domain of LRRK2. To test our hypothesis, we examined whether Prx2 overexpression could influence the phosphorylation statue of Rab10, a well-studied LRRK2 substrate[22]. As shown inFig. 2D, Prx2 overexpression could significantly decrease the phosphorylation of Rab10 compared to the control,while Prx2 itself had no noticeable inhibition on the basal level of Rab10 phosphorylation (data not shown), strongly supporting our hypothesis that Prx2 might be an upstream inhibitor of LRRK2 kinase activity.
Prx2 involves in LRRK2 induced apoptosis
Previous studies have demonstrated that LRRK2G2019Scould significantly increase phosphorylation of Prx3, which was associated with decreased peroxidase activity and increased death in LRRK2 overexpressed neuronal cells[17]. However, we have proven that Prx2 has a higher affinity than Prx3 in interacting with LRRK2, and Prx2 overexpression could decrease phosphorylation of its substrate. Thus,we determined whether Prx2 is associated with LRRK2 cellular functions.
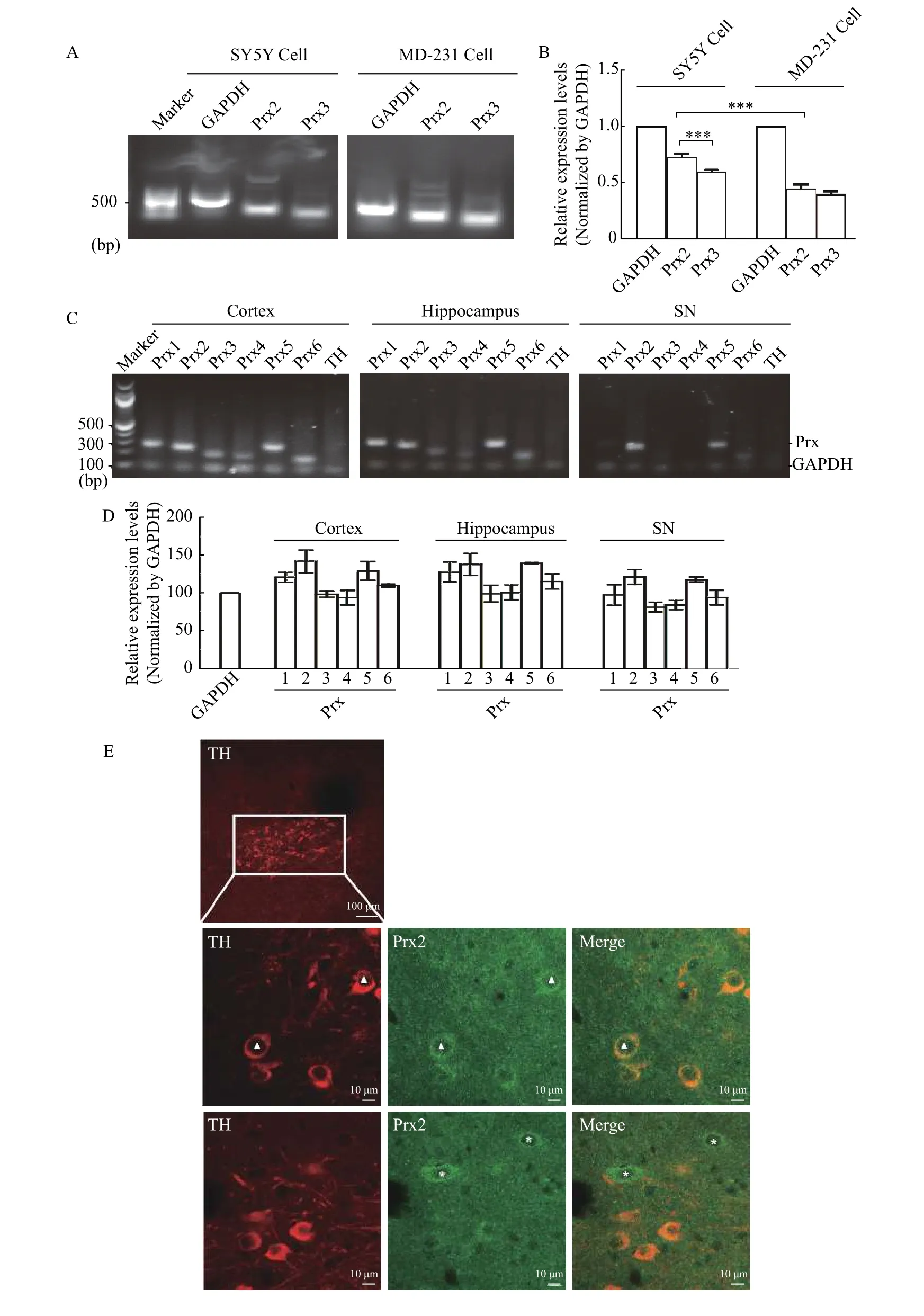
Fig. 1 Prx2 is preferentially expressed in both neuronal cell line and dopaminergic brain regions. A: The mRNA levels of Prx2 and Prx3 were examined in SH-SY5Y and MDA-MB-231 cells by RT-PCR. PCR products were separated by the 1% agarose gel. GAPDH was used as internal control. B: Data of mRNA levels were presented as mean±SEM from three independent experiments. Significance was determined using the one-way ANOVA. ***P<0.01. The results showed that the expression level of Prx2 was significantly higher in neural cellderived SH-SY5Y cells than in non-neuronal cells. C: The mRNA levels of Prx1-6 isoforms in different brain regions were examined by RTPCR. PCR products were separated by the 1.7% agarose gel. GAPDH was used as internal control and TH was used as indicator of dopaminergic neurons. D: mRNA levels were presented as mean±SEM from three independent experiments. The results showed that the expression level of Prx2 was significantly higher among Prxs, especially in SN region. E: Immunofluorescent staining to detect the specific localization of Prx2 in dopaminergic neurons was performed using anti-Prx2 antibody and anti-TH in rat SN regions. Prx2 (Green) was mainly present in cell bodies and Prx2-positive cells were colocalized with TH (Red) in cell bodies. Scale bar, 10 μm.
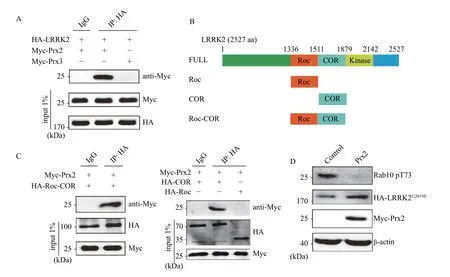
Fig. 2 LRRK2 specifically interacted with Prx2. A: 3HA-LRRK2 was co-transfected with Myc-Prx2 or Myc-Prx3 in COS-7 cells, followed by cell lysates co-immunoprecipitated (co-IP) with anti-HA antibody and then analyzed with anti-Myc antibody using Western blotting. B: Schematic representation of LRRK2 constructs used in this study. C: Myc-Prx2 was co-transfected with different LRRK2 truncated constructs (Roc-COR, Roc, COR), followed by cell lysates co-IP with anti-HA antibody and then analyzed with anti-Myc antibody using Western blotting. D: 3HA-LRRK2G2019S or 3HA-LRRK2G2019S and Myc-Prx2WT were transfected in HEK-293 cells. After 24 hours, the level of Rab10 pT73 in the cells was measured by the tag antibody of Rab10 pT73 to indicate the inhibitory role of cotransfected Prx2 on LRRK2G2019S kinase activity.
Given the presence of multiple enzymatic domains,LRRK2 dysfunction has been associated with a diverse set of cellular functions including autophagy,apoptosis and retrograde trafficking[23]. First, to test whether Prx2 could influence the autophagic activity caused by overexpressing LRRK2G2019S, we cotransfected LRRK2G2019Swith Prx2 wild-type (WT),T89E mutant which showed phosphor-mimic of Prx2,and T89A mutant which exhibited resistance to phosphorylation. The levels of LC3-II were examined as the indicator of autophagy induced by starvation.As shown inFig. 3A, LRRK2G2019Sdramatically increased the level of LC3-II under nutrient-deprived condition, which is consistent with previous studies,indicating that PD-associated LRRK2 mutant could induce autophagy. Surprisingly, overexpression of Prx2 WT, T89E, and T89A did not reverse the abnormally increased LC3-II caused by LRRK2G2019S,which is not consistent with the effect of Prx3 on autophagy. This result implied that Prx2 and Prx3 might play different roles in their interaction with LRRK2.
Next, we deployed Terminal deoxynucleotidyl transferase (TdT) dUTP Nick-End Labeling (TUNEL)assay to explore whether Prx2 could rescue the LRRK2G2019Sinduced apoptosis. As shown inFig. 3BandC, the apoptotic signal was significantly increased in LRRK2G2019Soverexpression group while Prx2WToverexpression could reverse the apoptotic process,suggesting that Prx2 could alleviate cellular toxicity induced by mutant LRRK2, which might relate to the inhibitory effect of Prx2 imposed on elevated LRRK2 kinase toxicity.
Reverse of altered retrograde trafficking induced by LRRK2G2019S by overexpressed Prx2
In addition to apoptotic toxicity, LRRK2 has been reported to play a role in the retrograde trafficking pathway revealed by altered retrograde trafficking of cargo proteins, such as CI-M6PR, from endosomes totrans-Golgi network (TGN)[24]. To test whether Prx2 took part in LRRK2 mediated retrograde trafficking pathway, we firstly examined the subcellular distribution of CI-M6PR using immunofluorescent staining. We found that both of overexpressed LRRK2WTand LRRK2G2019Sled to mis-targeting of CIM6PR from perinuclear pattern to diffused pattern,while co-transfect with Prx2 could partly rescue this altered subcellular localization (Fig. 4A). Since overexpressing LRRK2G2019Swould alter retrograde trafficking of CI-M6PR and cause a distinctly decreased half-life of CI-M6PR, presumably due to increased targeting to lysosomal pathway, we then examined whether co-transfected Prx2 would increase CI-M6PR half-life. Compared with LRRK2G2019Soverexpression, the half-life of CI-M6PR was significantly decreased in Prx2 KD cells while it was rescued in Prx2-overexpressed cells (Fig. 4CandD).The knockdown efficiency was tested by Western blotting (Fig. 4B). These combined data based on the subcellular localization and protein stability studies indicated that Prx2 could alleviate the altered retrograde trafficking caused by LRRK2G2019Soverexpression.

Fig. 3 Effect of Prx2 on autophagy and apoptosis induced by LRRK2 G2019S. A: HeLa cells were transiently overexpressed 3HALRRK2G2019S with Myc-Prx2WT or Prx2 mutations (Myc-Prx2T89A and Myc-Prx2T89E). After 24 hours, cells were treated with DMEM to induce starvation for 2.5 hours followed by cell lysates analyzed endogenous LC3 A/B using Western blotting. B–C: In HeLa cells, 3HALRRK2G2019S was transferred separately, or 3HA-LRRK2G2019S and Myc-Prx2WT were transferred together. After 24 hours, fixed cells were stained using the TUNEL kit. FITC labeled the apoptotic cells (green fluorescent) while DAPI indicated cell nuclear. Scale bar, 50 μm. Data from three independent experiments were presented as mean±SEM. One-way ANOVA was used to analyze the significance between groups.***P<0.01.
Discussion
The reported work here was based on a novel LRRK2 binding protein Prx2 from a yeast two-hybrid screen. We showed that Prx2 was preferentially expressed in TH neurons which is more relevant to PD related tissue specificity. We further confirmed that Prx2 specifically interacted with the COR domain and inhibited the kinase activity of LRRK2 which may be intramolecularly regulated through the Roc-GTPase.Finally, we showed that Prx2 rescued PD mutant LRRK2 induced apoptosis and reversed the altered retrograde trafficking of TGN protein CI-M6PR,suggesting the inhibitory function of Prx2 in LRRK2 mediated PD pathogenesis.

Fig. 4 Inhibitory role of Prx2 on altered retrograde trafficking of CI-M6PR induced by LRRK2. A: HeLa cells transiently overexpressed GFP-hCI-M6PR with LRRK2WT or LRRK2G2019S, or combined with Prx2 were double immunofluorescent stained to examine the subcellular distribution of the chimeric receptor protein. Scale bar, 10 μm. B: HeLa cells were transiently transfected with scrambled or specific Prx2 siRNA for 24 hours. Prx2 protein level in the cell lysates was detected with anti-Prx2 endogenous antibody by Western blotting.C: Half-life of HA-hCI-M6PR-tail expression was examined in control and Prx2 depleted HeLa cells with overexpressed LRRK2G2019S, or Prx2 and LRRK2G2019S after CHX (100 μg/mL) treatment on the indicated time points (0, 6, and 12 hours). D: HA-hCI-M6PR-tail expression level was detected with anti-HA antibody by Western blotting and quantified by densitometry analysis. E: Relative CI-M6PR level was presented from three independent experiments. One-way ANOVA was used to analyze the significance between groups. ***P<0.01.
Relevance of interaction of Prx2 with LRRK2 in PD pathogenesis
By using the pheochromocytoma PC cell origin cDNA library in the Y2H screening[25], we aimed at finding regulatory interactors of LRRK2-Roc-COR domain that has some dopaminergic secretory phenotype[26]. With the identification of Prx2, we further showed its relevance to PD pathogenesis by determining its expression in dopaminergic neurons compared to that of Prx3 whose interaction with LRRK2 was also recently reported[17]. As shown inFig. 1, among all known members of Prx family,Prx1, 2 and 5 were highly expressed in the cortex and hippocampus of rat brain. Interestingly, only 1 and 5 were preferentially expressed in the SN which is enriched in dopamine neurons whose selective loss leads to PD related movement disorders. The cellular staining also verified the association of Prx2 with TH positive neurons in SN (Fig. 1). While the neuronal association of Prx2 in the brain is consistent with literature report[14,27]and compensatory increased expression of Prx2 in PD[28–29], the preferential expression of Prx2 over the Prx3 in both SY5Y cell and SN supports the relevance of Prx2 in its interaction with LRRK2. Furthermore, the specific interaction analysis by co-IP and domain dependence of this interaction (Fig. 2) suggested that Prx2 and Prx3 interact with LRRK2 differently. Certainly, the relative high expression of Prx3 in neuronal cells may be also associated with its functional relevance of its interaction with LRRK2. Specifically, Prx2 has a higher affinity in binding to full length of LRRK2 than Prx3 (Fig. 2). And their binding domain requirement is COR for Prx2 in our study and the kinase region for Prx3 binding[17]. Thus, whether LRRK2 binds to both Prx proteins to perform synergetic neuroprotective function remains to be investigated.
Prx2 inhibits the kinase activity of LRRK2 through interacting with COR domain
As we started to characterize the nature of the interaction, we assumed that Prx2 may also serve as another potential substrate for LRRK2 kinase activity,similar to Prx3[17]. However, our functional analysis showed neither mutant forms of Prx2 (T89A and T89E) played different roles from that of the wild type(Fig. 3and data not shown). By using Rab10 as substrate for LRRK2 kinase, the co-expressed Prx2 strongly inhibited the LRRK2 induced Rab10 phosphorylation (Fig. 2). Further functional assay such as apoptosis and membrane trafficking also supported such notion that Prx2 might be the upstream of LRRK2 kinase activity that is quite different from Prx3. A plausible explanation for how Prx2 regulates LRRK2 kinase activity may be related to its interaction to COR domain. COR domain is believed to be a dimerization device responsible for Roc-GTPase activity[30]. Other studies also indicated that COR domain interacts with Roc which in turn regulates its activity[31]. Interestingly, recent studies have indicated that there is intramolecular GTPasekinase regulation within LRRK2[32]. The GTP binding to Roc-GTPase is required for active kinase activity and neurotoxicity of LRRK2 in PD pathogenesis[4,21,31,33]. Thus, we suggest that, through binding to COR domain, Prx2 may interfere with the function of COR in dimerizing Roc-GTPase with reduced activity which further inhibits the kinase activity and the cellular toxicity of LRRK2.
Inhibitory role of Prx2 in mutant LRRK2 induced cellular toxicity
Prxs are Cys-based peroxidases that highly abundant and conserved in tissues from bacteria to humans. They play crucial roles in protecting cells from oxidative stress, and maintaining genome stability and longevity. Although how Prxs are related to autophagic process are not clear, Prxs were detected on the membrane of autophagosome[34]and can be processed after ROS induced ubiquitylation by autophagy[35]. Further studies indicated that the antioxidative function of Prx2 may depend on the types of cell and intracellular condition[36]. That Prx2 overexpression alone as well as coexpression with LRRK2 failed to inhibit starvation induced autophagy in our data may suggest that autophagy recruit Prx2 instead of inhibiting its further interaction with LRRK2. On the other hand, LRRK2 induced apoptosis can be rescued by overexpressed Prx2. One explanation is that Prx3 is involved in mitochondria related process while Prx2 participated by different mechanisms[17]. Furthermore, Prx3 serves as the substrate of LRRK2 kinase activity to alter its antioxidative activity as a feedback process[17], while Prx2 acts as upstream inhibitor of LRRK2 that may alleviate the LRRK2 toxicity. Additionally, as LRRK2 plays a role in altering membrane trafficking through phosphorylating a set of small GTPase Rabs such as Rab29, Rab10,etc.[37–38], the neuroprotective role of Prx2 in LRRK2 toxicity may be mediated by blocking the altered retrograde trafficking of LRRK2 as indicated previously[24]. Thus, subcellular distribution and protein stability of CI-M6PR through retrograde trafficking were indicators for Prx2 mediated protective role in mutant LRRK2 kinase induced pathogenic toxicity in cells.
Acknowledgments
This work was supported by the National Nature Science Foundation of China to Y. Liu (Grant No.31371436 and No. 8157051134), and the laboratory start-up grant from Nanjing Medical University to Y.Liu. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
杂志排行

THE JOURNAL OF BIOMEDICAL RESEARCH的其它文章
- The current status of malignant hyperthermia
- Contribution of neutrophils in the pathogenesis of rheumatoid arthritis
- H2S protects against diabetes-accelerated atherosclerosis by preventing the activation of NLRP3 inflammasome
- AAV-mediated human CNGB3 restores cone function in an allcone mouse model of CNGB3 achromatopsia
- The level of bile salt-stimulated lipase in the milk of Chinese women and its association with maternal BMI
- RNA-seq analysis identified hormone-related genes associated with prognosis of triple negative breast cancer