De novo assembly of the seed transcriptome and search for potential EST-SSR markers for an endangered, economically important tree species: Elaeagnus mollis Diels
2020-05-19YulinLiuSiqiaoLiYunyingWangPingyuLiuWenjingHan
Yulin Liu · Siqiao Li · Yunying Wang · Pingyu Liu · Wenjing Han
Abstract Seeds of Elaeagnus mollis (Elaeagnaceae) produce an edible oil and contain more vitamin E (Ve) than major oil-seed crops. Despite its economic value, there is no information on its genome sequence. Here,we used the Illumina platform to determine the seed transcriptome of E. mollis to identify the genes related to Ve biosynthesis and potential simple sequence repeat (SSR) markers. In total, 100,999 unigenes were obtained with an average length of 605 bp and N50 of 985 bp. Of these unigenes,52,256 (51.7%) were annotated in at least one public database (NT,NR,PFAM,SwissProt,KOG,KO,and GO)in searches using blastn/x. The unigene annotation identified 15 unigenes encoding six enzymes (GGR, HPPD,HPT/VTE2, MPBQ-MT/VTE3, TC/VTE1, and γ-TMT/VTE4)putatively involved in Ve biosynthesis.In addition,16,810 SSRs distributed in 14,057 unigenes were mined.Of these,2820,583,and 3423 SSRs were located in the 5′-UTR, coding sequence (CDS), and 3′-UTR regions,respectively, while the remaining 9984 SSRs had undetermined physical locations. The largest group of repeat motifs comprised mononucleotide repeats (70.76%), followed by dinucleotide(15.59%)and trinucleotide(12.10%)repeats. AG/CT (8.69%) and AAG/CTT (4.15%) were the main dinucleotide and trinucleotide repeats, respectively.Furthermore, 9597 SSR-specific primer pairs were designed. Among 100 primer pairs selected randomly to determine their usefulness,53 proved to be efficient.To the our best of knowledge, this work is the first study of the E. mollis transcriptome and constitutes valuable genomics data for future genetic engineering studies to alter the amount of Ve. The identified potential EST-SSR markers can be used for population genetics studies and assistedbreeding of E. mollis.
Keywords Elaeagnaceae · Elaeagnus mollis ·Transcriptome · Vitamin E · Simple sequence repeats(SSR)
Introduction
Vitamin E (Ve) compounds are tocochromanols, which include tocopherols and tocotrienols. Tocopherols and tocotrienols have four isoforms (α-, β-, γ-, and δ-tocopherols/tocotrienols), and all are potent lipophilic antioxidants that can be synthesised only by photosynthetic organisms (Me`ne-Saffrané and Pellaud 2017). In humans and animals, Ve is essential for reproduction and is a highly effective lipid antioxidant that prevents damage by free radicals in cell membranes.Thus,Ve may also reduce the risk of certain cancers and cardiovascular diseases(Chen et al. 2006). Because the main source of dietary Ve is plants, increasing the Ve content in the edible parts of plants,especially in seeds that are rich in oil would help Ve dietary needs.
Elaeagnus mollis Diels (Elaeagnaceae) is a unique Palaeozoic survivor of the Quaternary glaciation and categorised as endangered in the International Union for Conservation of Nature Red List (Version 2018-2, http://www.iucnredlist.org). In the wild, it grows only in Shanxi and Shaanxi Provinces, China (Zhang and Zhang 2015;Wang et al.2017a).During the past decade,it has attained the status of an ecologically and economically important forest tree species and has been cultivated widely because it is an excellent honey plant and its leaves make a healthy tea (Wang et al. 2017b). More importantly, the seeds of E. mollis are highly nutritional, containing large amounts of a high-quality edible oil, Ve (119.6-126.6 mg/100 g in seed oil), and many essential amino acids and trace elements (Liang et al.2015). Therefore,identifying the genes involved in Ve synthesis and accumulation in E. mollis could help increase the output of Ve with gene engineering techniques.
Recently, numerous studies on E. mollis have focused on population genetics and evolution (Wang et al. 2012;Zhang and Zhang 2015), physicochemistry of the nut oil(Liang et al.2015;Kan et al.2017),and tissue culture(Yan et al. 2004). However, few studies have attempted to identify the genes involved in Ve biosynthesis and develop useful molecular markers,partly because of the scarcity of genomic information.
Simple sequence repeat(SSR)markers have been used for population genetics studies, association mapping and marker-assisted selection in many crops species because of their abundance, co-dominant inheritance, highly polymorphic nature, and random distribution throughout the genome (Wang et al. 2016a). There are two basic types: genomic SSRs and expressed sequence tag (EST)SSRs. Compared with genomic SSRs isolated from random genomic sequences, EST SSRs, which are located in the coding regions of functional genes, are more likely to be linked to important agronomic characters (Gailing et al.2013).With the use of RNA-sequencing technology,many EST-SSR markers have been developed for plant species such as Neolitsea sericea (Chen et al. 2015b),
Hevea brasiliensis (Li et al. 2012) and Rhododendron latoucheae (Xing et al. 2017). In comparison, only a few SSR markers have been identified for E. mollis using traditional protocols (Wang et al. 2012; Ye et al. 2016).These markers are insufficient for population genetics studies or marker-assisted breeding in E. mollis. Therefore, the development of SSR markers in E. mollis is necessary.
The main objectives of this study were to(1)generate a valuable genetic resource for discovering candidate genes to enhance targeted agronomic traits such as the Ve content of seed oil in other plants by genetic engineering and (2)identify SSR loci, design EST-SSR primers, and examine the potential of EST-SSRs for marker development,genetic diversity studies, and molecular breeding programs of E. mollis.
Materials and methods
Plant materials and transcriptome sequencing
Fresh seeds (90 days after flowering) of E. mollis were obtained at the Arboretum of Northwest A&F University,Shaanxi Province, China. The seeds were immediately snap-frozen and stored in liquid nitrogen for RNA extraction. Total RNA was extracted using TRIzol reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s protocol. The quality and quantity of total RNA were assessed using the RNA Nano 6000 Assay Kit of the Agilent Bioanalyzer 2100 system (Agilent Technologies, CA, USA) and a NanoPhotometer spectrophotometer(IMPLEN,Westlake Village,CA,USA).For RNA sample preparation, 1.5 μg RNA was used as input material. Sequencing libraries were generated using NEBNext Ultra RNA Library Prep Kit for Illumina (NEB, Ipswich,MA, USA) and the manufacturer’s recommendations.Illumina sequencing was done using the Illumina HiSeq 2000 platform at Beijing Novogene Biological Information Technology Co., Ltd., Beijing, China (http://www.novogene.com/).
De novo assembly, functional annotation,and classification
First, raw reads were processed using in-house perl script to remove adaptor, poly-N, and low-quality reads to generate clean reads. Then, the clean reads were used for de novo assembly using Trinity software with the default parameters to obtain transcripts (Grabherr et al. 2011).Through the assembly of components from the potential alternatively spliced transcripts, the longest transcripts extracted from each cluster of units were defined as unigenes.
All unigenes were compared against public protein and nucleotide databases, such as NCBI non-redundant protein sequences (NR), SwissProt, Protein Family (PFAM),Eukaryotic Orthologous Groups of Proteins(KOG),KEGG Orthology database (KO), and Gene Ontology (GO) via blastx and NT(NCBI non-redundant nucleotide sequences)using blastn (E-value <10-5). Based on the NR annotation, GO terms were obtained using Blast2GO v2.5.0 software (Götz et al. 2008). Then, the program WEGO(http://wego.genomics.org.cn) was used to describe the distributions of GO terms. In addition, the statistical enrichment of genes in the Kyoto Encyclopaedia of Genes and Genomes (KEGG) pathways was determined using KOBAS software (Mao et al. 2005).
Coding sequence (CDS) prediction
The prediction of CDSs in the unigenes was done in two steps: first using blastx to search the NR and SwissProt protein databases, and then using EST Scan software(3.0.3)(Iseli et al.1999)to predict a CDS if a unigene had no specific blastx matches in these two databases. The E-value thresholds of the two steps were both set to less than 10-5.
SSR detection, primer design and validation
SSRs were mined in all unigenes using the MISA tool(http://pgrc.ipk-gatersleben.de/misa). To identify the mono-, di-, tri-, tetra-, penta-, and hexanucleotide motifs,the parameters were set to a minimum number of 12, 6, 5,5, 4, and 4 repeat units, respectively. Two or more SSRs separated by less than 100 bp were defined as compound SSRs. The physical locations of the SSRs were assigned according to the predicted 5′-untranslated regions (5′-UTRs), CDS, and 3′-untranslated regions (3′-UTRs). On the basis of the MISA results, Primer3 software (http://primer3.sourceforge.net) was used to design primer pairs for each SSR locus with the following criteria: (1) length ranging from 18 to 27 bp,(2)size of PCR products ranging from 100 to 280 bp, and (3) annealing temperature set at 57-62 °C.
To validate the usefulness of these primer pairs, we randomly selected 100 pairs for synthesis by Shanghai Sangon Biological Engineering Technology (Shanghai, China) and tested each pair in a PCR reaction with 5 μL (10 ng/μL) of template DNA, 1.5 μL of 2 μM each primer, 10 μL of 2 × Taq Master Mix (Dye Plus, Vazyme Biotech; Nanjing,China), and 2 μL of ddH2ofor total volume of 20 μL. The PCR amplification profile consisted of 95 °C for 5 min; 35 cycles of 94 °C for 30 s,55 °C for 30 s,and 72 °C for 40 s;and a final 10-min extension at 72 °C. The PCR products were separated by electrophoresis on 1% agarose gels, and their sizes were estimated against a DNA ladder.
Results and discussion
Sequencing and de novo assembly
Compared with Sanger sequencing, next-generation sequencing technology is a cost-effective way to obtain large numbers of gene sequences, especially for nonmodel organisms (Cao and Zhang 2017). In this work, the Illumina sequencing platform produced 55,233,650 raw reads,which were deposited in the NCBI Sequence Read Archive(SRA) under accession number SRP120068. After the reads with adaptor sequences and low-quality reads were removed, 53,630,830 clean reads were obtained. After de novo assembly with the program Trinity, 135,784 transcripts were generated with a mean length of 812 bp.After the longest transcript in each transcript cluster was then extracted, 100,999 unigenes were obtained with a mean length of 605 bp and N50 of 985 bp. Of the unigenes,29,769 (29.47%) were greater than 500 bp long (Table 1).
Functional annotation and compare with related species
To learn more about the features and functions of these unigenes, we used them in a search against public databases, including NT, NR, SwissProt, PFAM, GO, KOG,and KO,using blastn/x(E-value <10-5).Of the unigenes,41,665 (41.25%), 30,564 (30.26%), 16,802 (16.63%),33,582 (33.24%), 29,165 (28.87%), 29,636 (29.34%), and 16,314 (16.15%) had significant matches in the NR, NT,KO, SwissProt, PFAM, GO, and KOG databases, respectively.Overall,52,256(51.74%)unigenes had a significant match in at least one database, while 7316 (7.24%) unigenes were annotated in all seven databases(Table 2).The remaining 48,743 (48.26%) unigenes (87.19% ≤500 bp and 1.54%>1000 bp) failed to generate a blast match in any of the databases (Fig. 1a). Because E. mollis is a unique Palaeozoic plant, these unannotated unigenes,especially those longer than 500 bp, might be new genes.
In the blastx search of the NR database, 41,568(41.16%)unigenes were found to have high similarity with sequences from Prunus mume (4649, 11.18%), Prunus persica(4103,9.87%),Vitis vinifera(3202,7.70%),Citrus sinensis (2383, 5.73%), Malus pumila (2152, 5.18%), and

Table 1 Characteristics of assembled transcripts and unigenes
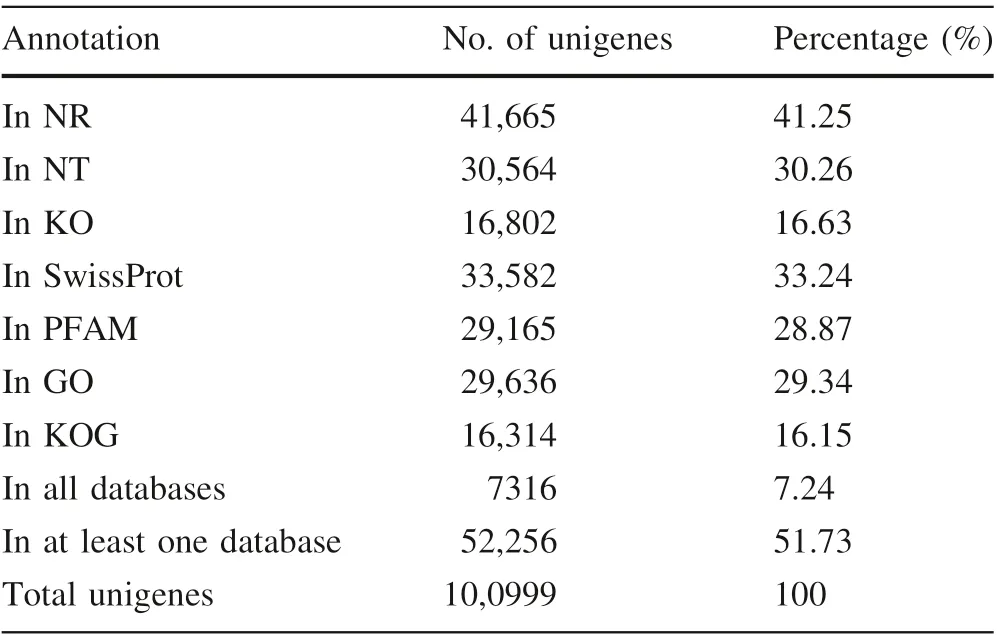
Table 2 Summary of functional annotation of assembled unigenes
other species (25,079, 60.33%) (Fig. 1B). From a phylogenetic perspective, P. mume, P. persica, M. pumila, and V.vinifera are all in the Rosidae clade(Chase et al.2016).Therefore, it is reasonable to speculate that E. mollis,which is also the root in the Rosidae lineage, will have a similar transcriptome composition in conservative regions.This confirmed that the transcriptome resource of E.mollis obtained here is accurate and effective for molecular biology research.
Functional classification of unigenes
For functional prediction and classification, all unigenes were assessed for GO assignments, KOG classifications,and KEGG pathway assignments.Overall,based on the NR annotations, 29,636 unigenes were categorised into 56 functional groups among three main GO categories: biological processes, cellular components, and molecular functions. As shown in Fig. 2a, within these functional groups, ‘‘binding’’ was the dominant group in molecular function,which included 15,577 unigenes(52.56%),follow by ‘‘catalytic activity’’ (13,328, 44.97%) and ‘‘transporter activity’’ (1983, 6.69%). In biological processes, ‘‘cellular process’’contained the largest number of unigenes(15,948,53.81%), followed by ‘‘metabolic process’’ (15,794,53.29%)and‘‘single-organism process’’(12,055,53.29%).The three large groups in cellular components were‘‘cell’’,‘‘cell part’’, and ‘‘organelle’’, which contained 9089(30.67%), 9083 (30.65%), and 6052 (20.42%) unigenes,respectively.
Of the 100,999 unigenes, 16,314 unigenes with significant matches were assigned to 25 KOG molecular families.The dominant groups were ‘‘general function prediction only’’ (2812, 17.24%), ‘‘post-translational modification,protein turnover,chaperones’’(2227,13.65%),and‘‘signal transduction mechanisms’’(1501,9.20%),while‘‘unnamed protein’’ was the smallest group, and included a single unigene (Fig. 2b).
In a search against the KEGG database,16,802 unigenes with significant matches were classified into 32 KEGG pathways affiliated with five main categories: cellular processes (A), environmental information processing (B),genetic information processing (C), metabolism (D), and organismal systems (E). Of these 32 pathways, the most represented were ‘‘signal transduction’’ (1858, 11.06%),followed by carbohydrate metabolism (1794, 10.68%),translation (1604, 9.55%), and overview processing (1293,7.70%) (Fig. 2c). Undoubtedly, these data will be useful for future metabolic research on E. mollis.
CDS prediction
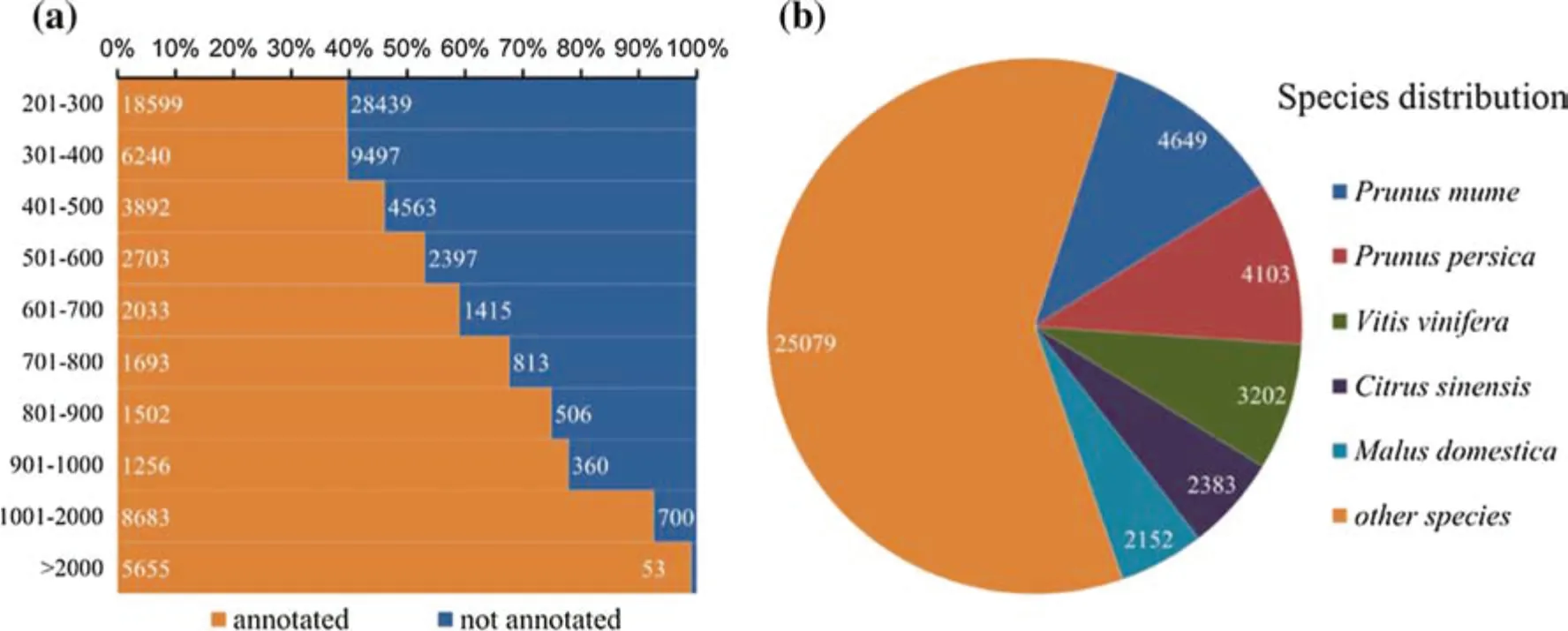
Fig. 1 Unigene functional annotation results. a Length distribution of annotated unigenes compared with those unannotated. b Top-hit species distribution for BLAST matches for the assembled unigenes
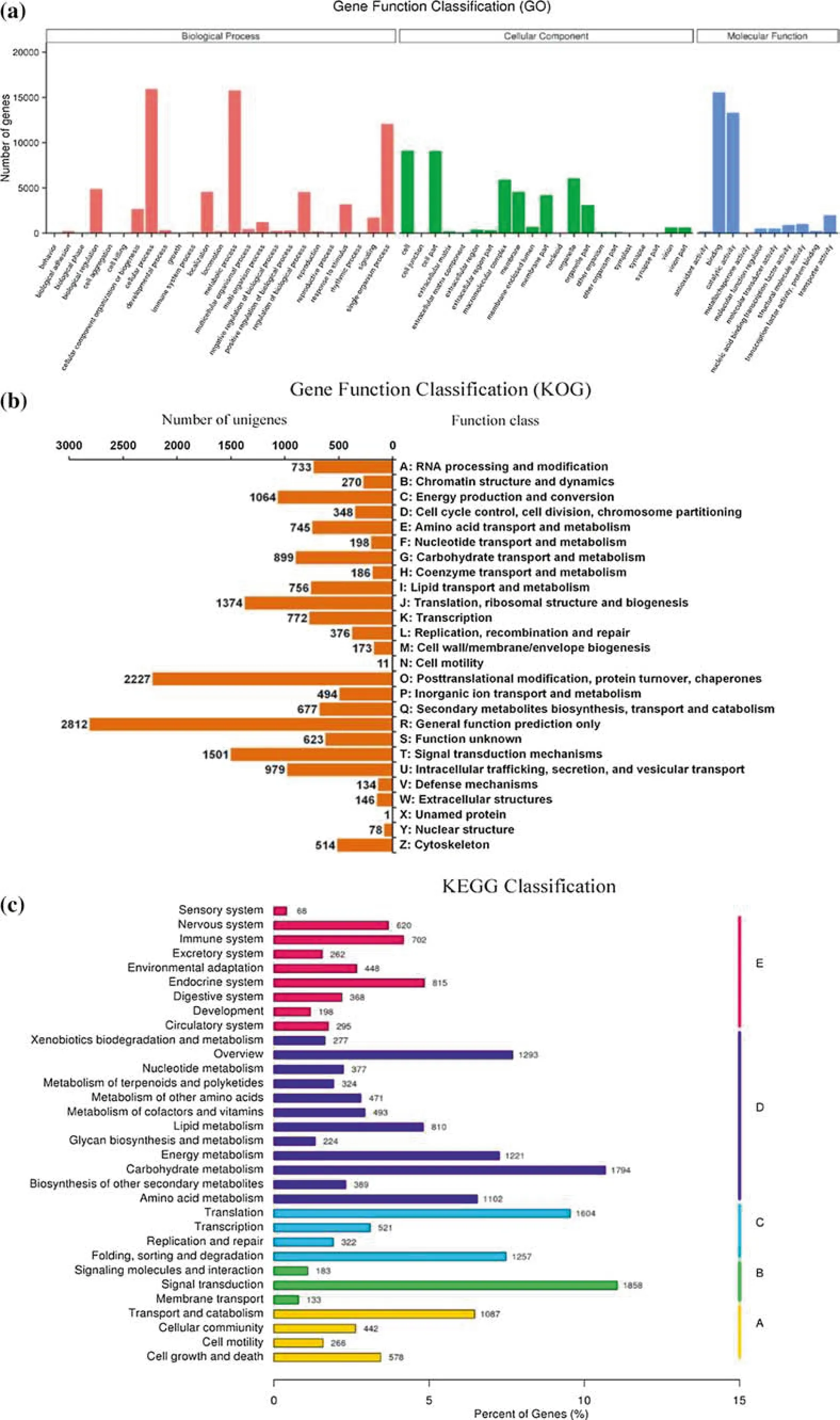
Fig. 2 Functional classification of unigenes by GO (a), KOG (b) and KEGG (c)
In all, 42,128 (41.71%) unigenes containing 45,201 CDSs were identified via blastx, and 30,684 (30.38%) unigenes containing 30,765 CDSs were translated into peptide sequences using EST Scan.However,the remaining 28,187(27.91%) unigenes had no CDS matches. Of the unigenes with CDS regions predicted by blastx, 20,851(49.49%)were longer than 500 bp and 13,140(31.19%)were longer than 1000 bp, which differed from the EST Scan predictions that the majority (24,641, 80.31%) were shorter than 500 bp. Moreover, 2875 (10.20%) unigenes were longer than 500 bp with no CDS matches(Fig. 3).The longer the unigenes were, the better the CDS regions could be predicted.
Identification of unigenes related to Ve biosynthesis
Most of the research on the biosynthetic pathways of Ve has focused on model plants(Harish et al.2013;Dorp et al.2015) and major oil crops such as soybean (Liu et al.2017b), canola (Du et al. 2015) and olive (Georgiadou et al.2015).The current work based on the KEGG pathway(ko00130) enrichment and unigene annotation identified four geranylgeranyl reductase (GGR), two 4-hydroxyphenylpyruvate dioxygenase (HPPD), one homogentisate phytyltransferase (HPT/VTE2), four 2-methyl-6-phytylbenzoquinone methyltransferase (MPBQ-MT/VTE3), two tocopherol cyclase (TC/VTE1), and two γ-tocopherol methyltransferase (γ-TMT/VTE4) unigenes related to Ve biosynthesis in the E. mollis seed transcriptome. Unfortunately, we did not find any unigenes encoding homogentisate geranylgeranyl transferase (HGGT), phytol kinase (VTE5), or phytyl-phosphate kinase (VTE6)(Fig. 4).
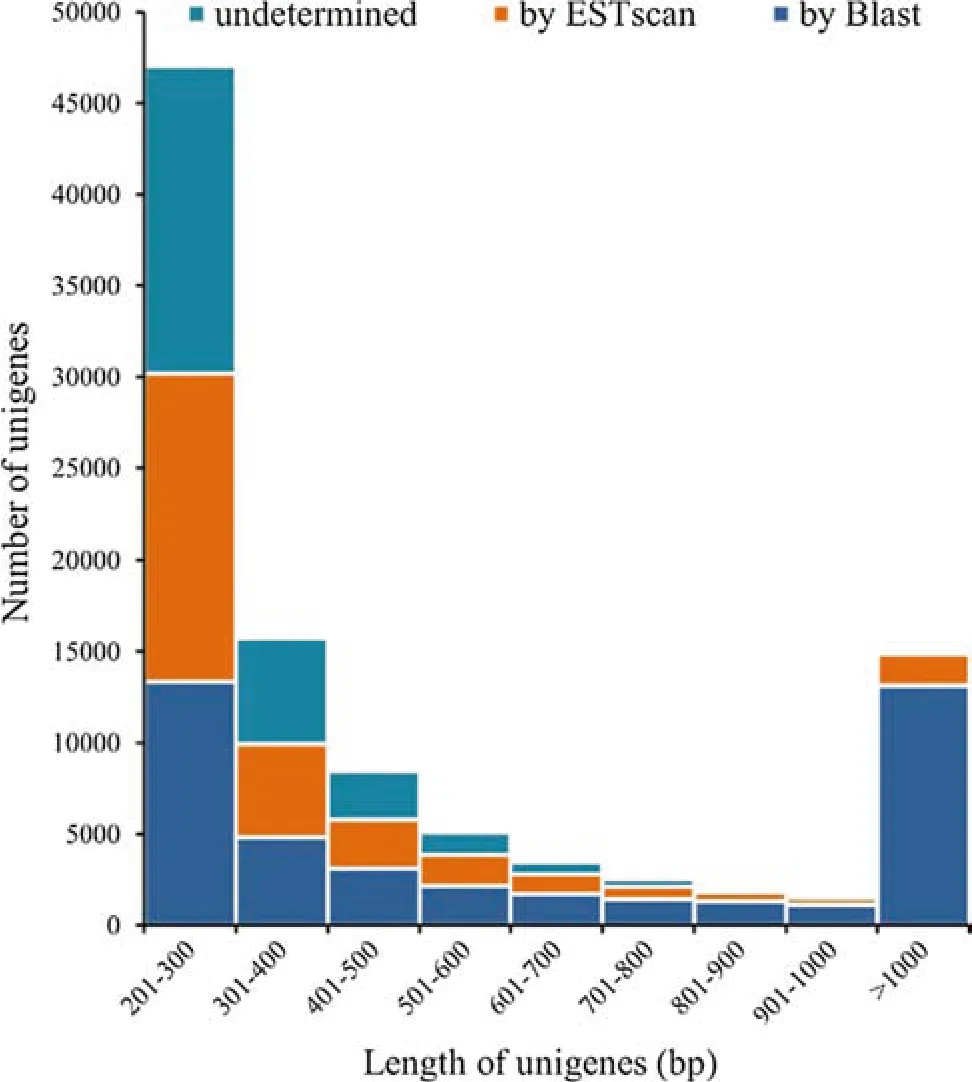
Fig. 3 Number of unigenes with CDS regions identified by BLASTx and EST Scan compared with those without CDS matches in different unigene length intervals
HGGT and HPT are two key enzymes related to the condensation of homogentisic acid(HGA)derived from the shikimate (SK) pathway with geranylgeranyl diphosphate(GGDP) and phytyl diphosphate (PDP) derived from the plastid methylerythritol phosphate (MEP) pathway to produce tocotrienols and tocopherols,respectively(Kong et al.2016). Tocotrienols are mainly found in monocot seeds,while tocopherols accumulate in almost all photosynthetic plants, especially in vegetable oils and seeds (Yang et al.2011).Therefore,we inferred that the expression of HGGT in the seeds of E. mollis was too low to be detected in this work. In Arabidopsis seeds and leaves, phytol from chlorophyll degradation is sequentially phosphorylated by VTE5 and VTE6 to produce phytyl monophosphate(PMP)and PDP, the substrate for tocopherol synthesis (Me`ne-Saffrané and Pellaud 2017). Because the seeds chosen for transcriptome sequencing in this study were approaching maturity, the presence of chlorophyll and phytol was unlikely and might be one of the main reasons that we did not find VTE5 and VTE6 in this transcriptome. Alternatively, the unigenes that encode HGGT, VTE5, and VTE6 might be too short to get a blast hit in the public databases.
Identification of SSRs
The great number of transcripts or unigenes generated by RNA sequencing provides sufficient sequence information to search for SSR loci and develop EST-SSR markers(Zhang et al. 2015). Of the 100,999 unigenes that we identified, 14,057 were found to contain 16,810 SSRs, and 2275 contained two or more SSRs. On average, there was one SSR locus in every 3.63 kb of the E. mollis transcriptome, which is denser than in the Sorbus pohuashanensis (Liu et al. 2017a) and Eremochloa ophiuroides transcriptome (Wang et al. 2016b). As shown in Table 3,the SSRs comprised 11,894 (70.76%) mononucleotide,2621 (15.59%) dinucleotide, 2034 (12.10%) trinucleotide,205 (1.22%) tetranucleotide, 29 (0.17%) pentanucleotide,and 27 (0.16%) hexanucleotide motifs. Among these, the most abundant motif detected in the SSRs was the A/T motif (11,727, 69.76%), followed by the GA/TC (1460,8.68%), AT/TA (880, 5.23%), AAG/CTT (697, 4.15%),AAT/ATT (396, 2.36%), CA/TG (277, 1.65%) and ATC/ATG(222,1.32%)motifs.The remaining motifs accounted for only 6.85%(1151)of the repeat motifs.This result was similar to those for S. pohuashanensis (Liu et al. 2017a),Fraxinus velutina (Yan et al. 2016) and Juglans mandshurica(Hu et al.2016).In addition,the physical positions of these SSRs in the unigenes were also identified, and 2820, 583, and 3423 SSRs were located in the 5′-UTR,CDS, and 3′-UTR regions, respectively. The locations of the remaining 9984 SSRs were undetermined because there was insufficient information to delimit the SSR loci in these unigenes to the CDS, 5′-UTR, or 3′-UTR regions.For the determined loci,most of the mono-,di-,tetra-,and pentanucleotide motifs were located in 5′-UTR and 3′-UTR regions, and most of the trinucleotide motifs were located in CDS regions, similar to the case in the Sargassum thunbergii transcriptome (Liu et al. 2016). Trinucleotide motifs (480, 82.33%) were the dominant type in the CDS regions, with AAG/CTT (161, 27.62%) the most common motif.
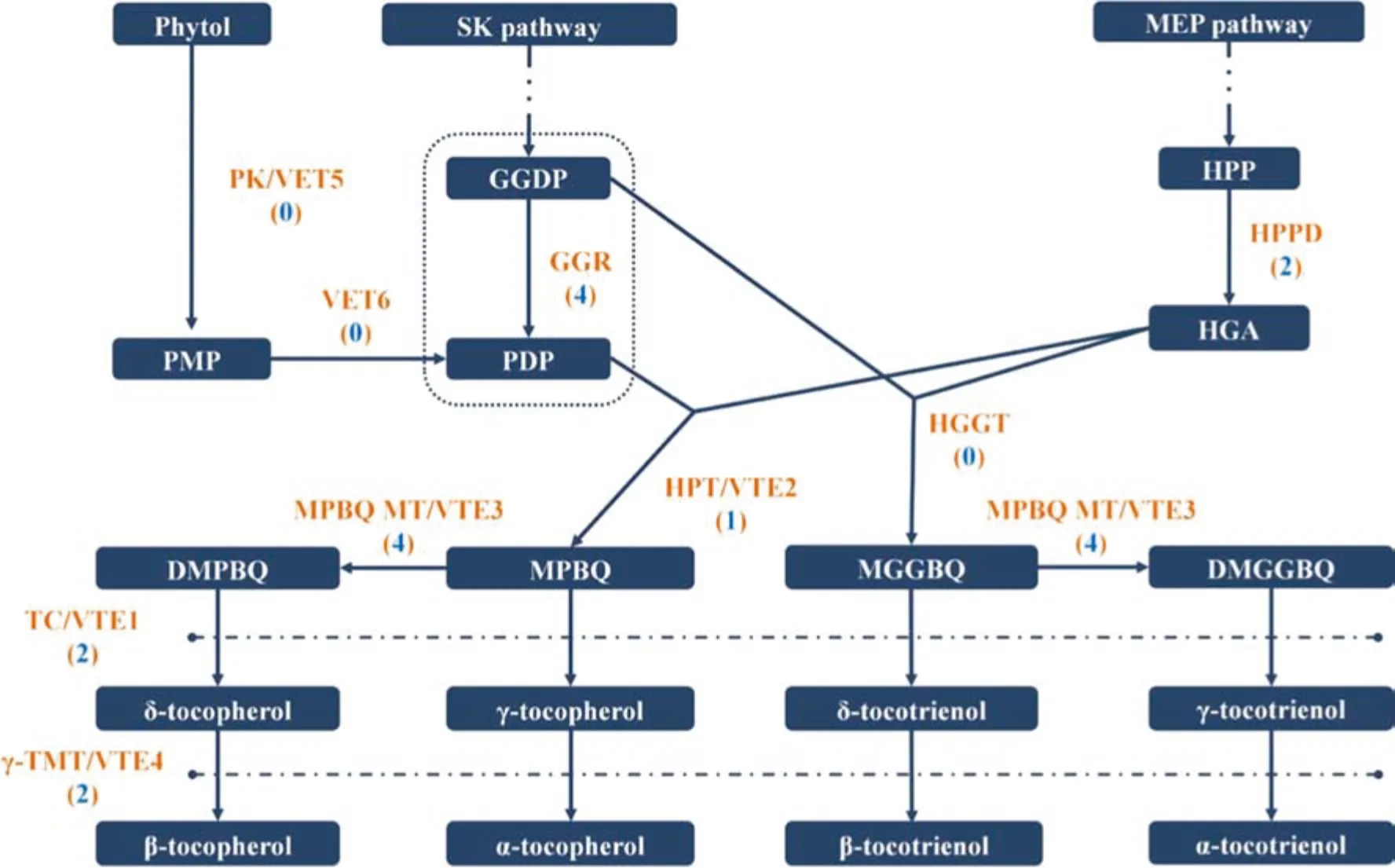
Fig. 4 Overview of tocopherol and tocotrienol biosynthesis pathways. Numbers shown in parentheses correspond to unigenes encoding enzymes in the tocopherol and tocotrienol biosynthesis pathways in E. mollis seeds. Substrate abbreviations: SK shikimate;MEP methyl erythritol phosphate; HPP p-hydroxyphenylpyruvate;HGA homogentisic acid; GGDP geranylgeranyl diphosphate; PMP phytyl monophosphate;PDP phytyl diphosphate;MPBQ 2-methyl-6-phytylbenzoquinone; DMPBQ 2,3-dimethyl-5-phytyl-1,4-benzoquinone; MGGBQ 2-methyl-6-geranylgeranylbenzoquinone; DMG GBQ 2,3-dimethyl-5-geranylgeranyl-1,4-benzoquinone. Enzyme abbreviations: PK phytol kinase; VTE6 phytyl-phosphate kinase;HGGT homogentisate geranylgeranyl transferase; HPPD hydroxyphenylpyruvate dioxygenase;HPT homogentisate phytyltransferase;MPBQ MT 2-methyl-6-phytylbenzoquinone methyltransferase; TC tocopherol/tocotrienol cyclase; γ-TMT γ-tocopherol/tocotrienol methyltransferase
SSR primer screening and verification
Primer pairs were designed successfully for 9597 EST-SSR loci using Primer 3, while the remaining loci may have been too short or had inappropriate flanking sequences for primer design. For evaluating the usefulness of these potential EST-SSR markers, 100 were chosen randomly and tested. In these tests, 79 primer pairs amplified reproducible products from the genomic DNA of E. mollis,whereas the remaining 21 primer pairs failed to amplify any PCR products at the annealing temperatures used. Of the 79 successful primer pairs, 22 pairs generated more than one band. These results suggest that E. mollis germplasm has high heterozygosity or that the primer specificity was not high enough. Four pairs yielded PCR products longer than the expected size,suggesting that the amplified regions exist in introns, as seen for Vigna radiate (Chen et al. 2015a) and Elymus sibiricus (Zhou et al. 2016). The remaining 53 primer pairs amplified target bands that can be used for further studies.
Conclusions
The first E. mollis transcriptome was obtained using the Illumina sequencing platform. In total, 100,999 unigenes were generated and 51.7% of them were annotated in the NT, NR, SwissProt, PFAM, KOG, KEGG, or GO databases. In addition, 16 unigenes that encoded six enzymes involved in Ve biosynthesis and 16,810 EST-SSR loci distributed in 14,057 unigenes were identified. Furthermore, 9597 EST-SSR-specific primer pairs were successfully designed. Marker validation revealed that 53 of the100 EST-SSRs amplified the expected products. This valuable sequence resource and potential EST-SSR markers will be useful for functional gene identification,genetic improvement, and marker-assisted breeding in E. mollis.
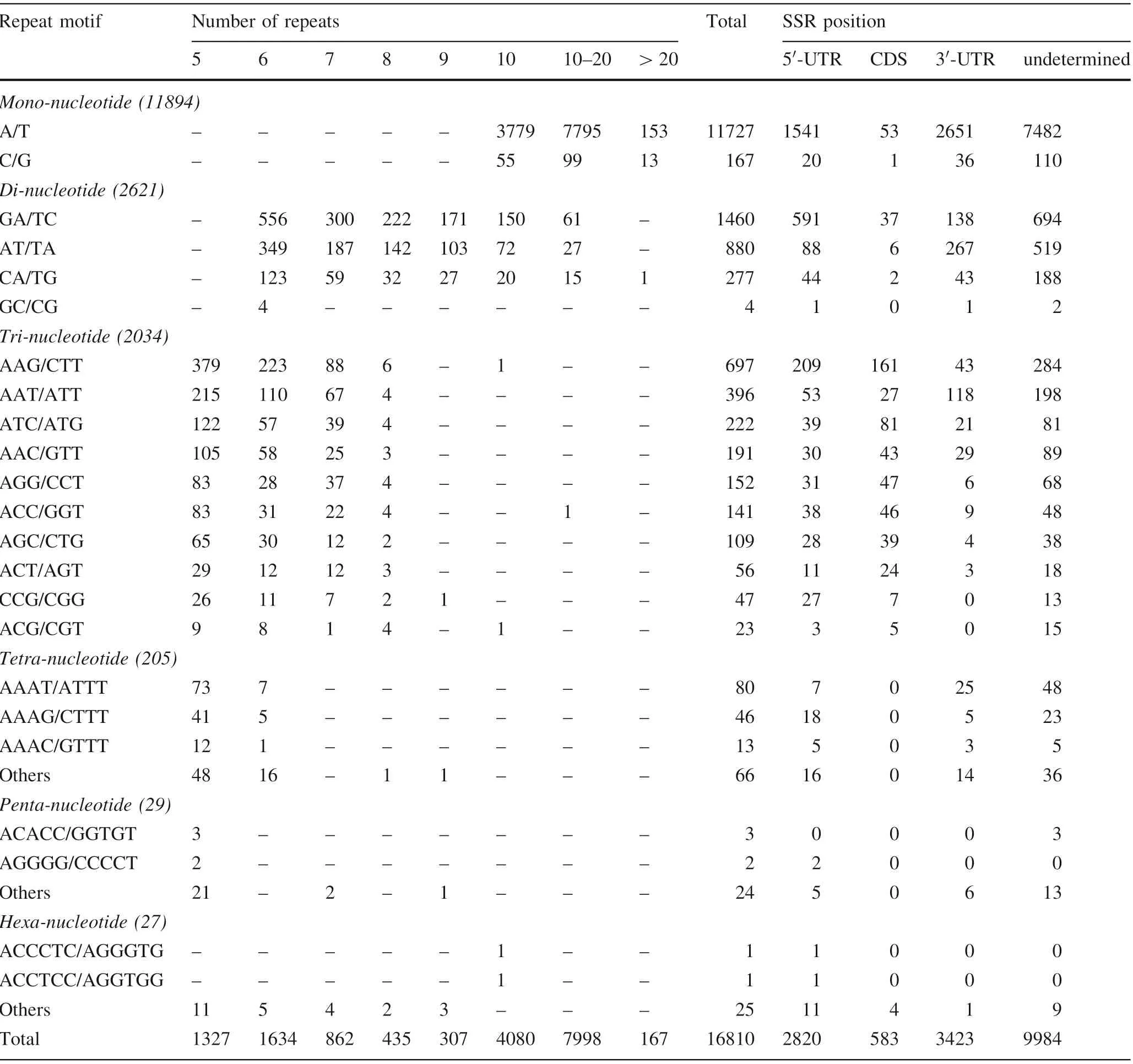
Table 3 Distribution pattern and predicted position of SSR motifs in the seed transcriptome of E. mollis
杂志排行

Journal of Forestry Research的其它文章
- Do increasing respiratory costs explain the decline with age of forest growth rate?
- At what carbon price forest cutting should stop
- Mapping the risk of winter storm damage using GIS-based fuzzy logic
- Effects of seed moisture content, stratification and sowing date on the germination of Corylus avellana seeds
- Comparison of seed morphology of two ginkgo cultivars
- Effect of artificially accelerated aging on the vigor of Metasequoia glyptostroboides seeds