三甲基转移酶SETD2表达下调促进结肠直肠癌细胞的迁移和增殖
2020-05-19丁家增陈海珍沈晓卉李超飞刘国梁
崔 昂, 丁家增, 陈海珍, 沈晓卉, 李超飞, 刘国梁
(上海交通大学医学院附属瑞金医院 外科/特需医疗保健中心,上海 200025)
近年来,微卫星不稳定(microsatellite instability,MSI)在结肠直肠癌(colorectal cancer,CRC)发病机制中的地位不断提升。曾有文献报道,电子结肠镜检查未发现明显异常的MSI表型病人,在36个月内发展成CRC[1]。近年有基因测序研究发现,高度MSI的CRC病人,SETD2的移码突变率高达6.7%[2]。TCGA数据库也显示,594例CRC病人有36例(6.06%)SETD2基因的位点发生改变,提示SETD2失活可能是导致体内MSI表型改变的潜在原因之一。本研究通过下调CRC细胞系SETD2表达,观察肿瘤细胞增殖、迁移能力的改变;通过裸鼠成瘤实验下调SETD2表达的肿瘤细胞,研究对肿瘤细胞成瘤能力的影响,和SETD2对CRC发生、发展过程的影响。
材料与方法
一、研究材料和RNA转染
取上海细胞研究所的CRC细胞系SW480及HCT116,培养增殖后用以转染。以pLKO.1为表达载体,构建短发夹 RNA(short hairpin RNA,shRNA)转染用质粒,干扰SW480及HCT116细胞系中SETD2的表达,分为对照组 (pLKO组)及KD组(sh1/sh2组)。并通过mRNA及蛋白质水平实验验证干扰效果。用SETD2 shRNA稳定转染细胞系进行细胞功能实验。
二、Western印迹实验
使用SETD2 shRNA稳定转染后的细胞系提取蛋白质,并通过SETD2及三甲基化H3K36抗体进行Western印迹实验,验证SETD2蛋白质水平变化及其催化产物三甲基化H3K36的蛋白质水平变化。
三、细胞增殖实验
胰酶消化,离心后重悬计数。CRC细胞稀释至 20 000个/mL,充分混匀,加至96孔板,保证细胞密度为2 000个/孔,分为对照组 (pLKO组)及KD组(sh1/sh2组)。每组设置3复孔,包括空白组。24 h后各孔内避光加入promega试剂20 μL/孔,暗箱内反应1 h。1 h后,使用酶标仪490 nm波长检测OD值,各孔与空白孔OD值之差为每孔细胞OD值。其后间隔24 h检测各孔OD值,观察不同组别OD值变化。共观察5 d。
四、细胞划痕实验
取各组对数生长期的稳定转染细胞,消化离心重悬,计数后分别接种8×l05个细胞/孔至6孔板。置于37℃、5%CO2细胞培养箱内培养。待细胞融合接近全满时,用无菌200 μL枪头在6孔板中央划一纵痕。PBS缓冲液轻柔洗去脱落细胞,更换培养基为低血清培养基,继续放入37℃、5%CO2培养箱内培养。36 h后显微镜下观察细胞向缺损中央迁移情况。根据0 h及36 h划痕宽度改变来判断细胞迁移速度及能力。
五、细胞迁移实验
细胞迁移实验是用胰酶消化处于对数生长期的细胞。离心后,1%低血清DMEM培养基重悬。计数后,将细胞稀释为1.2×106个/mL。在transwell上室(24 孔,孔径 8.0 μm)加入 100 μL 细胞悬液,在下室加入600 μL正常DMEM培养基(含 10%FBS)。 在 37 ℃、5%CO2孵箱中培养 12~16 h后。吸除培养基,PBS缓冲液洗涤,棉签擦去上室底膜上层表面的细胞。4%多聚甲醛固定15 min,PBS缓冲液洗涤,结晶紫37℃染色30 min。100倍光镜下,选取四周及中央共5个视野计数,其平均数反映各组细胞的迁移能力。
六、裸鼠成瘤实验
选取3周龄雄性裸鼠。实验前1 d准备SETD2 shRNA稳定转染的SW480和HCT116细胞系,铺板10 cm细胞皿,细胞密度在70%左右。实验当天,使用2.5%胰酶溶液消化细胞,使用2倍胰酶体积、含有10%胎牛血清的DMEM中和,1 000 r/min离心3 min,去除上清液。加入10%PBS缓冲液重悬,1 000 r/min离心3 min。加入少量10%PBS缓冲液重悬,放入无菌消毒离心管中备用。
计数细胞,使细胞密度在1 000万个/mL,取1.5 mL备用。行裸鼠两侧腋下皮下注射,每侧注射0.1 mL上述制备细胞液,即100万个肿瘤细胞。标记裸鼠,每天观察,记录肿瘤出现时间和肿瘤直径。裸鼠肿瘤长至平均直径1 cm时,处死裸鼠、取出肿瘤。
剪开裸鼠表皮,暴露肿瘤。尽可能充分游离肿瘤周围组织。取出肿瘤后称重,剪取肿瘤中心一小块组织。提取RNA分析,与对照组比较肿瘤组织SETD2的表达含量。剩余肿瘤浸泡于多聚甲醛溶液,包埋石蜡切片,使用三甲基化H3K36的抗体进行免疫组织化学抗体染色,研究组织中该蛋白质表达变化情况。
结 果
一、shRNA干扰SETD2的mRNA表达
HCT116和SW480转染SETD2 shRNA,旨在构建SETD2下调的细胞稳系。如图1所示,通过实时PCR的方法验证SETD2的mRNA表达水平。结果表明,2个shRNA序列(sh1/sh2)在2个细胞系中均使SETD2 mRNA表达明显低于对照组(pLKO组)(P<0.01)。
二、下调SETD2的细胞SETD2和三甲基化H3K36蛋白表达水平的变化
HCT116和SW480中SETD2的mRNA表达水平下调后,Western印迹检测发现SETD2蛋白表达明显下调(β-actin为参照物),其催化产物三甲基化H3K36蛋白表达水平在组蛋白3整体水平不变的情况下明显下调(见图2)。
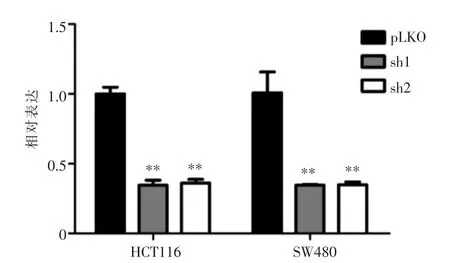
图1 转染SETD2 shRNA后HCT116和SW480细胞的SETD2 mRNA表达
三、细胞增殖实验
将HCT116、SW480中SETD2下调表达的统称为KD (knock down,sh1、sh2)组。与对照组相比,sh1、sh2 细胞增殖能力明显上升(P<0.05)(见图 3)。
四、细胞划痕和迁移能力实验
细胞划痕实验中,CRC细胞系SW480,SETD2表达下调后,细胞迁移能力明显提升。第2天,SETD2 KD组(sh1、sh2)划痕接近愈合,而对照组划痕清晰,表明KD组的迁移能力明显强于对照组(见图 4)。

图2 SETD2 shRNA转染后HCT116和SW480细胞系SETD2和三甲基化H3K36蛋白表达
transwell实验中,在100倍显微镜下,48 h后处理细胞,可见KD组结晶紫染色细胞明显多于对照组(见图5)。sh1组穿过transwell小室的细胞数在HCT116和SW480中分别是对照组的1.40倍和1.83倍。sh2组穿过transwell小室的细胞数分别为2.19 倍和 1.93 倍 (P<0.05)(见图 6), 表明 SETD2 KD组与对照组相比迁移能力明显增强。
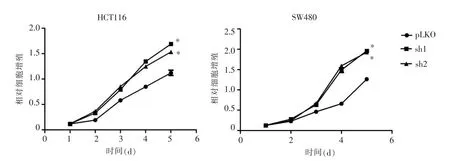
图3 SETD2 shRNA转染后HCT116和SW480细胞增殖变化

图4 SETD2 shRNA转染后SW480细胞系划痕实验
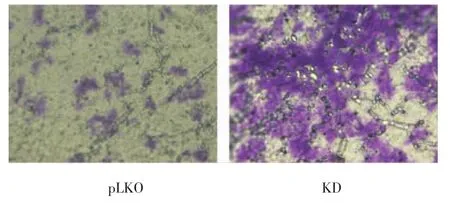
图5 SETD2 shRNA转染后HCT116 transwell实验结晶紫染色(×100倍)
五、SETD2表达对裸鼠成瘤的影响
使用SETD2表达下调的HCT116和SW480细胞系对裸鼠进行皮下注射,记录裸鼠肿瘤发生和生长天数。HCT116细胞系KD组在第11天出现肿瘤,对照组在第13天出现肿瘤。SW480细胞系KD组在第15天出现肿瘤,对照组在第21天出现肿瘤。KD组比对照组肿瘤发生的时间早,KD组肿瘤直径明显大于对照组(P<0.01)(见图7)。处死小鼠后,取出肿瘤称重,KD组重量明显大于对照组(P<0.01)(见图 8)。对裸鼠的肿瘤组织中心部分进行取样,提取肿瘤组织RNA。实时PCR检测肿瘤组织SETD2 mRNA表达水平,发现KD组裸鼠肿瘤SETD2表达明显低于对照组(P<0.05)(见图 9)。
六、裸鼠肿瘤组织三甲基化H3K36蛋白表达情况

图6 SETD2 shRNA转染后HCT116和SW480 transwell实验
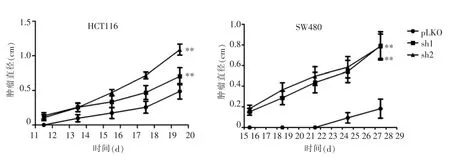
图7 裸鼠成瘤实验肿瘤直径
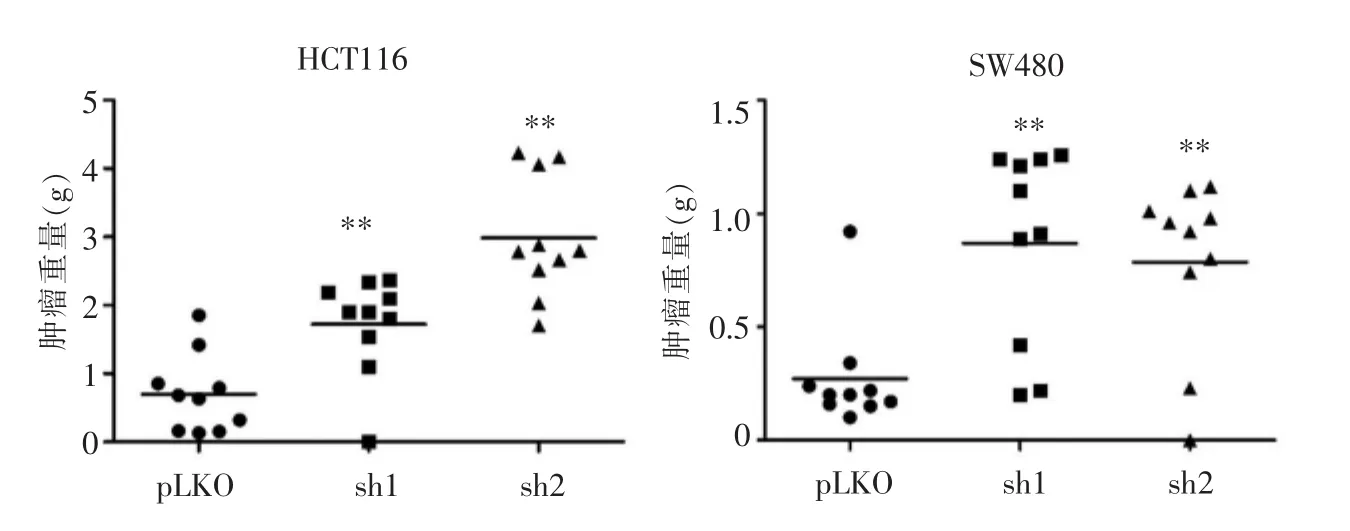
图8 裸鼠成瘤实验肿瘤重量
裸鼠肿瘤组织切片,并用三甲基化H3K36抗体进行免疫组织化学染色。40倍显微镜下观察三甲基化H3K36抗体染色的病理切片,发现对照组裸鼠肿瘤三甲基化H3K36蛋白表达呈强阳性(见图10A),KD组裸鼠肿瘤三甲基化H3K36蛋白表达明显低于对照组(见图10B)。图10C为阴性对照。
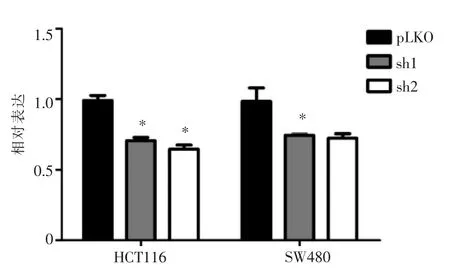
图9 裸鼠成瘤实验肿瘤组织SETD2 mRNA表达
讨 论
一、组蛋白H3K36三甲基转移酶SETD2
SETD2蛋白又名KMT3A或HYPB,其编码基因位于第3染色体3p21.31区域。SETD2蛋白是组蛋白H3K36特异性三甲基转移酶,相关分子量为230×103,含SET结构域,也被认为与亨廷顿病相关[3]。体内SETD2与左旋异构核糖核蛋白(hnRNPL)结合成复合物,行使酶的功能,且只影响三甲基化H3K36的表达水平[4]。SETD2的主要结构包含如下:①WW结构域和AWS-SET-postSET三联结构域,负责与H3K36特异性结合,发挥甲基转移酶活性;②一个保守的低电荷区域,富有谷氨酰胺和脯氨酸的转录激活结构域SETD2,通过羧基端与高度磷酸化的RNA聚合酶Ⅱ相结合[5],但与非磷酸化的RNA聚合酶Ⅱ不相关。SETD2可能是组蛋白甲基化修饰与基因转录调控之间的桥梁。随着对SETD2研究的不断深入,发现其在转录起始的抑制、RNA聚合酶Ⅱ转录延伸、DNA的修复、生物的发育成熟等过程中均发挥重要作用。
TCGA数据库显示,SETD2突变出现在多种肿瘤,包括CRC、散发的肾透明细胞癌、急性白血病、胃癌、肺癌、乳腺癌。SETD2突变导致肿瘤组织三甲基化H3K36表达明显下降。在某些肿瘤中,SETD2突变甚至与病人的预后、肿瘤对化疗的抵抗以及肿瘤的进展分期相关[4-6]。
二、CRC发生、发展过程中SETD2发挥抑癌作用
CRC发生、发展主要是以下两种途径:①染色体不稳定,即遵循“正常黏膜-腺瘤-腺癌”的规律,此途径最早的基因突变一般是APC基因,导致一系列抑癌基因如p53的失活。②DNA错配修复(mismatch repair,MMR)功能异常,DNA MMR功能缺失会导致一种特殊表型,即MSI。
(一)SETD2失活导致三甲基化H3K36表达降低而提高患癌风险
SETD2的羧基端与磷酸化的RNA聚合酶Ⅱ作用,参与基因的转录延伸过程[3]。SETD2既催化组蛋白H3K36三甲基化,又通过羧基端与RNA聚合酶Ⅱ相连,是组蛋白表观遗传学修饰与基因转录调控之间的重要桥梁。
哺乳动物SETD2是H3K36唯一的三甲基化转移酶,产物是三甲基化的组蛋白H3K36(H3K36me3)。SETD2表达下调,导致三甲基化的H3K36表达显著下降[9],并进一步发生DNA MMR功能缺失,从而表现出MSI的特征。在未检测到MMR基因突变的情况下,SETD2突变失活可能是发生MSI的原因。因此,SETD2被认为是潜在的抑癌基因。本研究CRC细胞系HCT116和SW480,通过shRNA干扰SETD2的表达 (见图1),发现在SETD2蛋白表达下调后,三甲基化H3K36在组蛋白3整体水平不变的情况下明显下调(见图2)。验证之前研究的结果,提示SETD2可能也是CRC的潜在抑癌基因。
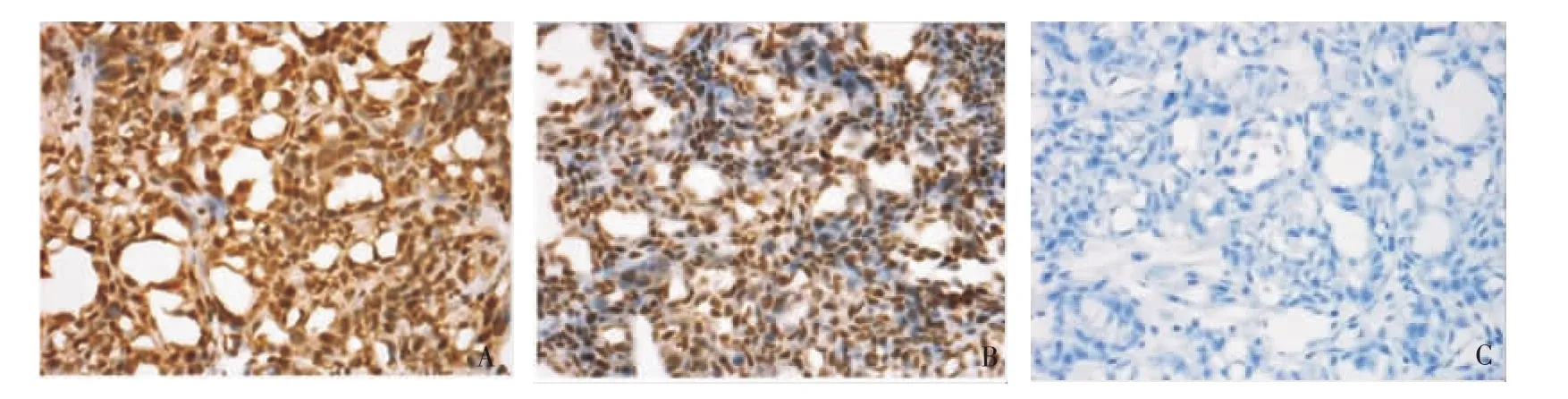
图10 免疫组织化学实验裸鼠肿瘤三甲基H3K36蛋白表达
(二)SETD2参与错配基因修复过程
近年来已有很多研究表明,肿瘤的发生与DNA的修复功能缺陷和细胞内异常转录活性升高密切相关。SETD2作为潜在的肿瘤抑制基因,编码的蛋白质对基因修复和抑制细胞内错误转录具有重要作用,是各种SETD2缺失肿瘤发生、发展的重要原因。
三甲基化H3K36对SETD2编码区去乙酰化,继而抑制转录起始的频率。SETD2突变导致三甲基化H3K36表达下降,体内错误转录起始过程明显增多[10],导致基因转录异常。SETD2与其他在转录延伸中恢复染色体正常结构的因子,如FACT复合物的亚基Spt6发生作用,抑制编码基因内隐藏的转录起始过程[11-12],保证基因转录的高保真度,从而防止肿瘤的发生。
人体MMR是体内重要的基因修复方式。MMR主要针对基因碱基配对错误和碱基插入丢失进行修复,是人体保证染色体稳定性的自然过程。MMR过程主要通过三甲基化H3K36识别hMutSα(hMSH2-hMSH6)和 hMutSβ(hMSH2-hMSH3)复合体来完成,其体内含量比值为10∶1。表观遗传学修饰对这些MMR基因(包括hMSH2、hMSH6)的影响会导致某些特定肿瘤的发生。在细胞层面,MMR缺陷导致特殊的突变表型,即MSI。因此MSI也被看作MMR缺陷的显著标志。研究表明,SETD2介导的H3K36三甲基化反应特异性结合hMutSα复合物,且有助于hMSH6在染色质内分布定位[13]。细胞内三甲基化H3K36含量G1期开始逐渐升高,S期早期达到峰值,S期末期和G2/M期最低,与hMSH6细胞内含量变化一致。进一步深入研究发现,MMR功能完整的人肺癌细胞系HeLa细胞中下调SETD2的表达,导致三甲基化H3K36表达也下调,且S期hMSH6含量比对照组显著下降,hMSH与三甲基化H3K36的共分布也比对照组显著减少,hMSH染色体定位发生明显改变。SETD2功能缺失导致三甲基化H3K36整体水平下降,特别是S期早期和G2/M期,从而影响细胞内MMR功能。SETD2失活突变的细胞表现出潜在MSI表型,随后细胞突变概率明显上升,最终导致肿瘤发生。Hela细胞中下调SETD2表达,显示出与DLD-1细胞系(缺乏hMSH6的MMR功能缺失的CRC细胞系)相似的 MSI状态[13]。
针对肾透明细胞癌、肺癌、胃癌和血液肿瘤的大规模测序均发现SETD2突变。SETD2缺失的肿瘤病人中,很多未发现MMR相关基因突变和失活。推断SETD2功能失活可能是MMR相关基因正常病人诱发肿瘤的重要原因[6,10,14]。
(三)SETD2参与调节p53转录因子和Wnt细胞通路
研究表明,SETD2选择性调控p53转录因子[15]。SETD2功能缺失导致p53无法被激活,从而增加肿瘤患病风险。小鼠结肠癌模型实验发现,SETD2失活起到关键作用[16]。在稳定的微环境中,SETD2对于正常生理并不是必需的。但一旦机体因辐射、疾病DNA受损、基因失活等影响内环境稳定,SETD2将会激活,并参与启动肠道上皮自我更新和修复,并随着SETD2在细胞内含量的下降,肿瘤发生的风险增加。深入研究证实,SETD2失活上调关键信号转导因子DVL2的表达,表明SETD2可能参与Wnt信号通路的调控,并最终影响肠道上皮重建和肿瘤发生、发展。
SETD2作为H3K36的三甲基转移酶,通过其产物三甲基化H3K36的表达变化,在CRC中引发MSI表型,并参与基因MMR过程。SETD2还作为调节因子调控p53、DLV2的表达,参与Wnt信号通路的调节,对CRC的发生、发展产生影响。本研究表明,SETD2下调抑制CRC细胞系 HCT116及SW480的三甲基化H3K36蛋白的表达,引起细胞功能的变化,导致肿瘤细胞增生及迁移更活跃。裸鼠成瘤实验中导致肿瘤生长更快,肿瘤的重量更大,提示SETD2在CRC中可能通过调控三甲基化H3K36的表达,参与DNA MMR修复过程,是CRC潜在的肿瘤相关基因,对CRC的发生、发展具有重要意义。
