PRiME HLB-UPLC-MS/MS法测定鲜蛋中6种药物残留
2020-05-18谭美龄
谭美龄 张 钦 付 鹏
(重庆市万州食品药品检验所 重庆 404000)
1 前言
鲜蛋因营养价值丰富,成为人们日常饮食中重要的动物源性食品之一,对其相关药物残留量的检测是食品安全监测的刚性需求。氯霉素类和喹诺酮类均为抗生素类药物,主要有氯霉素(Chloramphenicol,CAP)、氟苯尼考(Florfenicol,FF)、甲砜霉素(Thiamphnicol,TAP)、恩诺沙星(Enrofloxacin,ENR)、环丙沙星(Ciprofloxacin,CIP)等。五氯苯酚(Pentachlorophenol,PCP)及其钠盐为有机氯农药,常用作杀菌剂、除草剂和杀虫剂。氯霉素类药物可影响造血功能并导致贫血。喹诺酮类药物会影响胃肠道和心血管系统的健康。五氯苯酚通过水载体广泛扩散,影响生态安全并产生生物蓄积,可能对人类造成致畸、致癌、致突变和其他危害[1]。
目前,国内外相关检测方法中,大部分方法需要衍生化[2-4],采用传统的固相萃取柱进行净化,基质效应较难控制,使得灵敏度低,检测限高,存在假阳性风险,在复杂样品基质中的应用能力有限。本文在样品净化步骤中选用了对鲜蛋中的磷脂和色素类物质具有较好去除能力的Waters Oasis PRiME HLB柱[5,6],该新型小柱无须活化平衡、淋洗、洗脱,基质干扰物直接吸附于填料内,直接收集滤液待测,简化了操作步骤,节省了时间,降低了基质效应;同时,结合高效液相色谱-串联质谱(UPLC-MS/MS)法建立了高灵敏度、快速准确、安全经济的药物痕量分析法。
2 材料与方法
2.1 材料与试剂
标准物质:氯霉素(D015026)、五氯酚酸钠(170710,100 μg/mL)、氯霉素-D5(161228,100 μg/mL)、环丙沙星-D8(160414)(北京曼哈格);氟苯尼考(DRE-C 13665000)、甲砜霉素(DRE-C17457000)、五氯酚-13C6(C15970100)(Dr.Ehrenstorfer);恩诺沙星(TC180203-07)、环丙沙星(AL181105-06)(Stanford Chemicals);恩诺沙星-D5(272238)(WITEGA Laboratorien Berlin-Adlershof)。
主要试剂:甲醇、乙腈(德国Merck 公司)。
2.2 仪器与设备
AB Sciex QTRAP 4500 高效液相色谱-串联质谱仪(美国 AB SCIEX 公司);TG20WS 离心机(长沙湘智离心机仪器有限公司);KQ-800DE 超声波清洗器(昆山市超声仪器有限公司);DN-12W 氮吹仪(上海楚定分析仪器有限公司);IKA MS 3 digital 涡旋振荡器(德国 IKA 集团)。
2.3 实验方法
2.3.1 标准溶液的配制
取氯霉素、氟苯尼考、甲砜霉素、恩诺沙星、环丙沙星,用甲醇配制成混合标准工作溶液(100 ng/mL);取恩诺沙星-D5、环丙沙星-D8、五氯酚-13C6,用甲醇配制成混合内标工作溶液(1 μg/mL);按照最终定容浓度为 0.2、0.5、1.0、2.0、5.0、10.0、20.0 ng/mL,采用空白样品基质溶液进行稀释配制。
2.3.2 样品制备
取样:取鲜蛋,去掉外壳,使用匀浆机搅匀,放入干净的容器中,密封并标记。
提取:称取2 g 样品至50 mL 离心管内,加入混合内标工作溶液(1 μg/mL)50 μL,加入 80%乙腈水溶液(含0.2%甲酸)10 mL,涡旋混合1 min,超声10 min,8 000 r/min 离心 10 min(温度小于 5℃)。
净化:取2mL 上清液,加入到Water Oasis PRiME HLB(6 cc,200 mg)小柱上,使其自然下滴通过,取1 mL 收集到的滤液置于氮吹管中,于40℃下用氮气吹至近干。将残余物用初始流动相复溶并定容至1mL,过0.22 μm 微孔滤膜后用于UPLC-MS/MS 测定。
2.3.3 仪器分析
(1)色谱条件
色谱柱:InfinityLab Poroshell 120 EC-C18 2.1×100 mm 1.9-Micron;柱温:35℃;进样量:5 μL;流动相:A 乙腈、B 水(含 5 mmol/L 乙酸铵);流速:0.20 mL/min;梯度洗脱:0~2 min。流动相A 含量为10%并保持2~6 min;流动相A 从10%线性增加至90%,并保持至8 min;8~9 min,流动相 A 从 90%线性回至 10%,并保持至11 min。
(2)质谱条件
离子源:电喷雾型(ESI);电喷雾电压:5 500 V/-4 500 V;检测方式:多反应监测(MRM);辅助加热气:50 units;雾化气:50 units;反吹气:30 units;雾化器蒸发温度:550℃;扫描方式:正负离子同时切换,相关质谱参数详见表1。

表1 质谱参数
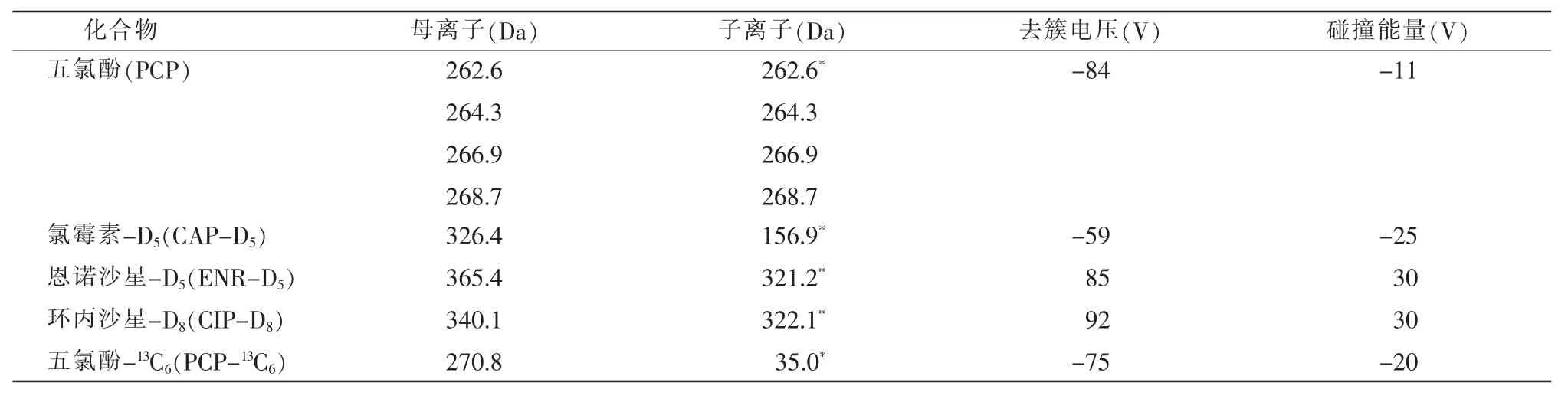
续表1
3 结果与分析
3.1 提取方法的优化
提取过程关系到整个检验工作的成败,6 种化合物中,除了五氯苯酚及其钠盐(PCP-Na)为农药外,其余5 种为兽药抗生素,均可溶于有机溶剂。根据这6 种化合物和鲜蛋基质的特性,本文分别选用甲醇、乙腈、乙酸乙酯作为提取溶剂。实验发现,3 种提取剂沉淀蛋白能力乙腈最好,其次为甲醇,产生的絮状沉淀易于冷冻离心分离。甲醇和乙酸乙酯能萃取出鲜蛋中的脂肪,而乙腈能减少脂肪和其他杂质的溶出,同时破除乳化现象。由于鲜蛋中含有大量水分,部分待测物如PCP-Na 易溶于水中,而乙酸乙酯只能溶解部分水分,不利于将目标物完全提取出来。所以本文选择与水混溶的乙腈作为提取溶剂,并加入部分极性溶剂水以形成混合提取体系,比较了提取液中乙腈体积分数分别为 100%、90%、80%、70%和60%时通过固相萃取柱时的提取效率(图1)。其中,80%的乙腈水提取的回收率最高且提取液更清亮易于后续的过柱净化。ENR 和CIP 含有羧基、哌嗪基,兼具弱酸弱碱的性质,在烯酸或稀碱中具有较好的溶解度。在酸性条件下PCP-Na 会酸化成脂溶性的PCP 而被提取出来,PCP 的 pKa=4.93,在酸性条件下形成分子态,对于增加其在有机溶剂中的分配比更有利。基于以上特性,本实验在80%乙腈水中添加0.2%的甲酸溶液,作为最终的提取溶剂。
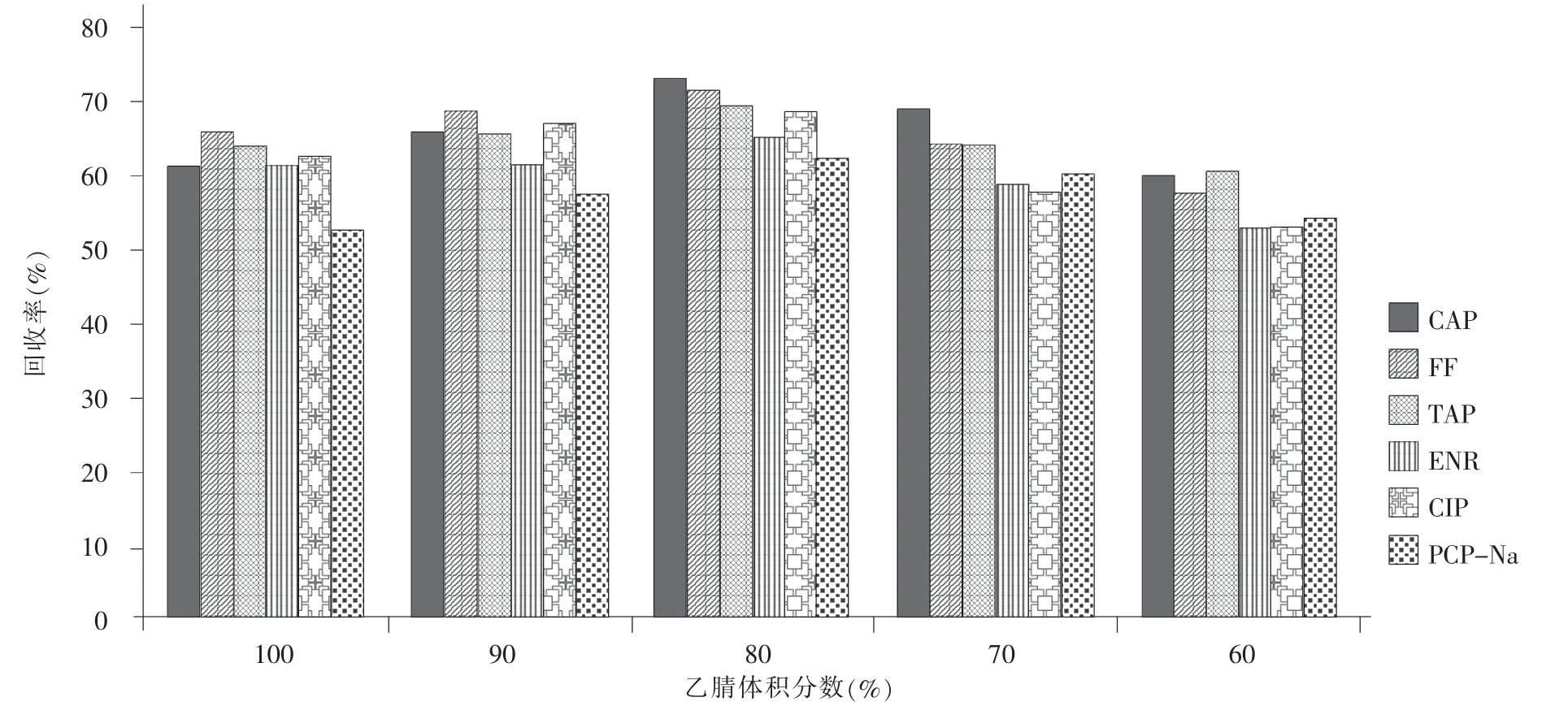
图1 不同比例提取液对净化效果的影响
3.2 净化方法的优化
鲜蛋的基质较为复杂,可能会干扰UPLC-MS/MS分析,影响定性定量。同时,还会污染色谱系统和质谱系统。所以,不仅要考虑目标化合物的提取效率,还要考虑在复杂的鲜蛋基质中的干扰物如脂肪、蛋白质、磷脂等的去除率。鲜蛋中的蛋白质通过乙腈沉淀和冷冻离心被去除。传统的溶剂脱脂步骤可以除去脂肪,但是有机溶剂消耗大,污染环境,不利于操作人员的健康。本实验采用固相萃取方法对样品进行净化,分别考察了 C18、HLB、PRiME HLB 3 种不同的固相萃取小柱的净化效果。其中,C18 的平均回收率只有70%~80%,HLB 和 PRiME HLB 均能达到80%~110%。通过质谱扫描不同小柱净化下的样品基质溶液,PRiME HLB 的基质干扰峰最少。C18 小柱是一种硅胶反相吸附剂,主要吸附疏水性目标物质,对脂溶性杂质如磷脂和色素,容易形成死体积使作用位点减少,并且小柱易干使目标物保留减少,净化能力有限。HLB 填料是亲水亲脂性单体的共聚物,具有高比表面积及强吸附性,也属于反相吸附机理,能够克服传统C18 填料的缺点,除杂能力与C18相比较好,但不能有效去除鲜蛋中的磷脂等干扰物。PRiME HLB 小柱是基于HLB 小柱的升级,同属反相保留机理,能有效去除鲜蛋中95%以上的基质干扰物,特别是对于脂肪、磷脂和色素的去除效果极好。同时,PRiME HLB 与 C18 和 HLB 小柱相比,无需任何活化、洗脱和淋洗,直接上样,使得样品净化过程更加简单、快速、有效。
3.3 液相色谱条件的优化
本实验选用具有较小填料内径(1.9 μm)的色谱柱对目标物进行分离,6 种药物均有较强的保留,分离较好。使用乙腈作为洗脱流动相,较甲醇的基线平稳,峰形更尖锐,响应更高。在ESI 电离模式中,挥发性的盐会堵塞取样孔,沾污透镜影响聚焦的电位分布,故本实验使用相对更易挥发的缓冲盐乙酸铵来提高目标化合物的电离效率。同时,采用梯度洗脱的方式,适当延长目标物的保留时间,将基质的干扰降到最低。由于ESI 质谱仪是浓度检测器,流速越小,在一定注射体积下流出峰的浓度越大,同时,低流速下产生的雾滴更利于雾化并增加信号强度。因此,使用0.2 mL/min 的流速并结合液相超高压技术的应用可以获得更好的峰形和灵敏度。
3.4 质谱分析条件的优化
据欧盟2002/657/EC 法案的要求[7],在农药和兽药残留分析中,对于禁用药物,以质谱法需要达到4 个点的要求。多级质谱的要求是检测1 个母离子和2 个产物离子,并分别用于定量定性。从质谱调谐可以看出,五氯酚结构非常稳定,其分子离子在碰撞室中不易被打碎,很难获得特征子离子。因此,选择五氯酚中氯元素的4 个同位素母离子作为质谱确认的识别点:262.6、264.3、266.9、268.7。其余 5 种兽药在碰撞室中均能被不同程度的打碎。通过全扫描方式观测离子情况,ENR 和CIP 易获得H+形成稳定的(M+H)+分子离子。CAP、FF、TAP 和 PCP 均为酸性化合物,容易丢失H+以形成稳定的(M-H)-分子离子,因此,本实验采用正负离子同时切换扫描对样品进行分析。在MS 定量分析时,采用了同位素内标物质,其中,CAP、FF、TAP 以 CAP-D5作为内标物,ENR 以 ENRD5作为内标物,CIP 以 CIP-D8作为内标物,PCP 以PCP-13C6作为内标物,以内标峰面积与目标物峰面积的比率进行定量分析,用于控制样品提取净化、LC进样、流动相和电离等过程的误差,提高定量的准确性。
3.5 基质效应的考察
在UPLC-MS/MS 中,由共流物引起的基质效应会影响分析方法的灵敏度、精密度和准确度。选取具有代表性的几种鲜蛋品种,通过对比溶剂标准曲线和空白基质工作曲线的差异,来考察不同鲜蛋基质对试验结果的影响。通过基质效应(Matrix Effect,ME)计算公式ME=[(基质匹配标准曲线斜率/纯溶剂标准曲线斜率)-1]×100%进行计算。当 ME<0 时,表现为基质抑制;当ME>0 时,表现为基质增强。不同鲜蛋样品的基质效应详见表2。由表2 可知,鹌鹑蛋为弱基质增强效应,鸡蛋、鸭蛋为中等强度基质增强效应,几种鲜蛋基质均有增强离子化的效果,所以需要配制基质匹配标准工作曲线来补偿基质效应。同时,本实验还通过稀释样品溶液、优化净化步骤、优化色谱分离条件共同作用来减少基质对实验结果的影响。
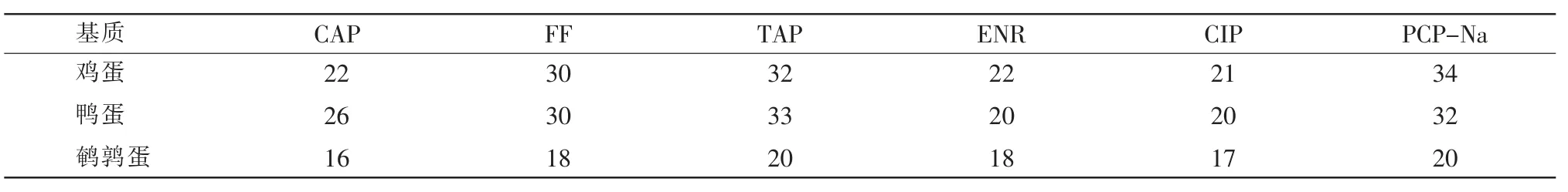
表2 不同鲜蛋样品的基质效应(单位:%)
3.6 方法学验证
3.6.1 线性范围及检出限
配制系列基质标准工作溶液,以峰面积对应的质量浓度做工作曲线,每个校准点以随机顺序重复测量2 次,结果详见表3。6 种化合物在质量浓度在0.2~20 ng/mL 时均具有良好的线性关系。符合GB/T 27417—2017《合格评定 化学分析方法确认和验证指南》[8]所规定的对于准确定量的方法,即线性回归方程的相关系数不小于0.99 的要求。

表3 工作曲线及检出限
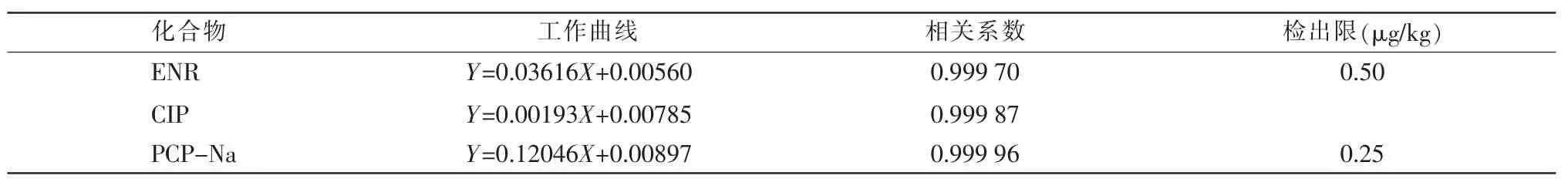
续表3
3.6.2 回收率及精密度
选用鲜蛋阴性基质样品,在曲线线性范围中做标准加入法,分别加入 2.5 μg/kg、5.0 μg/kg、25.0 μg/kg 3 个不同浓度梯度量的标准溶液,按照样品制备过程同法处理,进行回收率实验,每次测试重复6 次,CAP 的平均回收率为 98.7%~103.4%,RSD 为 1.6%~3.5%;FF 的平均回收率为 91.5%~95.8%,RSD 为 5.9%~7.8%;TAP 的平均回收率为87.4%~92.2%,RSD 为5.9%~7.3%;ENR 的平均回收率为90.2%~98.2%,RSD 为1.7%~3.5%;CIP 的平均回收率为 94.2%~96.2%,RSD 为1.9%~3.2%;PCP-Na 的平均回收率为 73.2%~75.4%,RSD 为3.1%~8.9%。以上数据表明,不同梯度的回收率均符合GB/T 27417—2017 附录A 规定的技术要求(浓度水平范围<0.1 mg/kg 时,回收率范围为60%~120%;精密度小于15%)。
4 结论
本文建立了PRiME HLB-UPLC-MS/MS 同时测定鲜蛋中6 种药物残留量的方法。结果表明,在本法条件下,优化了前处理,提高了准确度和精密度,6种药物在浓度为0.2~20 ng/mL 时线性良好,适合大批量的日常分析检测,可为建立同时快速测定兽药和农药残留的研究提供技术支持。
