蛋白质残基相互作用网络在线服务及可视化分析
2020-05-18管泽雨邱嘉迪刘文硕赵蕴杰
管泽雨,邱嘉迪,刘文硕,赵蕴杰
(华中师范大学物理科学与技术学院 生物物理研究所,武汉 430079)
蛋白质由20种不同的氨基酸经肽键聚合组成,通过形成特定的空间三级结构以实现催化和调控等不同的生物学功能.不同的氨基酸排列顺序会形成不同的蛋白质结构,蛋白质三维空间结构决定了其生物学功能[1-2].因此,蛋白质三维结构研究对理解其如何发挥生物学功能和设计相关的药物具有重要的意义.近年来,蛋白质残基相互作用网络普遍应用于蛋白质相关问题的研究.该方法中网络的节点为组成蛋白质的残基,网络的边为非共价键残基相互作用(如范德瓦尔斯和静电相互作用等)[3].基于蛋白质残基相互作用网络,可以进一步利用图论的方法研究蛋白质结构稳定性[4-5],蛋白质动力学[6-8],酶活性和变构调节[9],信号转导[10-11]等问题,为解决这些问题提供了一个崭新的视角.例如,Vendruscolo等人通过蛋白质残基相互作用网络的聚类系数(clustering coefficient),平均路径长度(average shortest path length)和中介中心度(betweenness centrality)的分析,提出了中介中心度极大值残基是折叠过程的关键氨基酸[8].Amitai等人通过计算蛋白质残基相互作用网络整体的接近中心度(closeness centrality)和相对溶剂可及性(relative solvent accessibility),可以较为有效地识别蛋白质活性位点氨基酸,在包含178个典型蛋白质的数据集中正确率达到70%[9].
目前已有一些分析蛋白质残基相互作用网络的工具.例如,AMINONET可计算疏水、亲水或带电氨基酸组成网络的拓扑属性[12];RING可构建残基相互作用网络,并通过Cytoscape[13]计算网络的拓扑性质[14-15];NAPS[16]可分析蛋白质网络边和节点的各种中心度性质,查找两个节点的最短路径,k连通子网等.然而,现有的蛋白质残基相互作用网络分析工具易用性较差,且网站访问较不稳定.例如,AMINONET需要在Java环境下运行,RING则需要将结果导出到Cytoscape进行分析,而Cytoscape本身也需要在Java环境下运行,需要依赖其它软件和环境,使用不方便.另一方面,随着人类基因组计划的完成,蛋白组学迅猛发展,大量的疾病相关基因被发现,药物作用的靶标分子急剧增加,现有方法缺乏对蛋白质结合口袋的分析,极大限制了残基相互作用网络方法的应用范围[17].因此,亟需易用性较强并可分析结合口袋的蛋白质残基相互作用网络模型.
本文建立了基于D3[18]和NGL Viewer[19]的蛋白质分析平台.用户仅需提交蛋白质的PDB结构信息,既可快速搭建蛋白质残基相互作用网络,计算网络的拓扑性质,实现网络的可视化,并结合DogSiteScorer计算的结合口袋信息计算它们的拓扑性质,对理解蛋白质的结构、生物学功能和相关药物设计有重要的意义.
1 算法
1.1 蛋白质残基相互作用网络
本文用蛋白质的三维结构信息构建蛋白质残基相互作用网络.网络由节点和边两部分组成:蛋白质结构中的氨基酸为网络中的节点.蛋白质结构统计分析表明,不相邻的氨基酸之间主要有两个距离分布的峰值.第一个距离峰值为0.3~0.5 nm,主要是氢键等短程相互作用;第二个距离峰值为0.7~0.8 nm,主要为静电等长程相互作用[20].因此,蛋白质相互作用研究大多数以0.8 nm作为距离截断,如果两氨基酸间任一对原子的距离小于0.8 nm则定义该氨基酸—氨基酸形成网络的边.
1.2 网络特征计算
本文提供了3类网络特征的计算,分别为度中心度,接近中心度和中介中心度,具体的计算公式如表1所示.度中心度直观上反映了一个节点在网络中的重要程度,定义为与该节点连接的边的数目;接近中心度描述了网络中的节点通过网络到达其它节点的难易程度,数值为该节点到其它所有能到达节点的平均距离的倒数,反映了该节点对其它节点施加影响的能力;中介中心度描述了节点在整个网络中的作用和影响力,定义为网络中所有最短路径经过该节点的数量[21].

表1 3种centrality的定义Tab.1 The definitions of 3 kinds of centrality
1.3 网站服务的基本框架
蛋白质残基相互作用网络服务用到了HTML、Javascript、PHP、MATLAB等语言以及D3.js、NGL.js等Javascript库,基本框架如图1所示.
主要流程为:
1) 服务器端调用FileRead.php,将临时文件传入到服务器的Inputs文件夹,对文件解压.
2) 调用Matlab编写的可执行程序,读取pdb文件中的数据,生成每个残基的3种centrality数据,具体为(流程图见图1(b)):①将pdb文件中的蛋白质三维结构数据转换为残基网络的邻接矩阵,若其中两个残基不相邻且存在一对原子距离小于0.8 nm则定义为相邻.②按照1.2中centrality的定义计算出每个残基的3种centrality值.
3) 清空Inputs文件夹,以便再次传入数据.将得到的每个残基的centrality值及残基之间的连接信息写入Outputs文件夹中的centrality.json文件,生成3种centrality的直方图,折线图,散点图.
4) 浏览器加载centrality.json文件,默认以closeness的倍数为节点的半径绘制所上传蛋白质的力导向图,并调用NGL Viewer显示蛋白质三维结构,显示3种centrality的直方图,折线图,散点图.
1.4 D3和NGL Viewer的实现
D3是一个基于web标准的JavaScript可视化库,D3可以借助SVG,Canvas以及HTML将数据生动的展现出来[18].使用基于D3的力导向算法(Forced-Directed Algorithm)的相关API来实现可视化网络.力导向算法是Eades于1984年提出的一种布点作图算法[22],其基本思想为将网络看成一个顶点为钢环、边为弹簧的物理系统,系统被赋予某个初始状态后,弹簧弹力的作用使钢环运动,直到系统总能量达到最小值时停止.通过改进力学模型可以得到不同的算法,并实现以下3点:1)节点分布均匀;2)边交叉最小化;3)具有对称性[23].此外,还通过添加tick事件来不断更新图形系统,实现动态的推拽效果.
NGL Viewer是实现蛋白质结构可视化的Web应用程序,用户可上传并显示蛋白质三维结构,不受第三方插件(如Flash和Java小程序)的影响.NGL Viewer支持常见的蛋白质结构文件格式(如pdb、mmcif)和各种分子表示(例如cartoon、spacefill、licorice、surface).此外,NGL Viewer可嵌入到网站中实现可视化.
2 结果分析
2.1 蛋白质残基相互作用网络在线服务的主要功能
研究表明,蛋白质残基相互作用网络呈现小世界网络特征[8,24],力导向图在小世界网络中有良好的可视化效果[25-26].力导向图易于理解和实现,可以画出相当优美的图形布局,充分展现出图的整体结构及其自同构特征[26].因此,基于力导向的可视化算法可以较好的描绘蛋白质残基相互作用网络.图2为细胞周期蛋白依赖激酶2(CDK2,PDB code:1fin,A链)前100个氨基酸的蛋白质残基相互作用网络,其中不同的5种颜色分别对应5个不同的口袋,黑色为默认颜色,表示不参与口袋的形成,右侧显示为该蛋白质的三维结构.

图2 细胞周期蛋白依赖激酶A链前100个氨基酸的力导向图Fig.2 Force-directed graph of top 100 amino acid of CDK2
网站的主要功能有以下几个方面.
1) 当光标浮在节点上方时,显示该节点的编号,closeness centrality,betweenness centrality,degree centrality,所在口袋编号(可能同时属于多个口袋);
2) 按照各centrality值高低搜索对应残基并染色;
3) 搜索某个口袋的所有残基并染色,观察其在网络中的分布;
4) 列出各个口袋的各平均centrality值;
5) 利用NGL Viewer显示蛋白质三维结构,并根据计算的centrality分布将三维结构染色;
6) 由D3绘制的力导向图具有拖拽功能,可以动态显示蛋白质残基相互作用网络;
7) 作出各centrality的区间分布图,centrality随氨基酸索引的折线图,不同centrality分别作横纵坐标的散点图.
2.2 蛋白质残基相互作用网络在线服务的使用方法
在线服务分为任务区,演示区和教程区,使用的主要步骤如下:
1) 上传蛋白质PDB结构文件,DoGSiteScorer(https://proteins.plus/#dogsite)计算得到的口袋结构信息(压缩为zip格式),提交任务.
2) 计算结果如图3所示,主要分为两个部分,区域I显示蛋白质的力导向图,区域II显示相应的三维结构图,初始时默认染色第一个口袋.此时,有两种染色功能可供选择:①根据centrality值的大小染色,centrality有closeness centrality、betweenness centrality、degree centrality三种,可在区域I下拉列表中选择.在文本框中输入整数m(下标从0开始)与n,点击color后突出显示排名在m与n之间的残基,将鼠标悬停在某个节点的上方,可以查看该节点对应残基的序号,各centrality值以及该氨基酸所在的口袋编号.②根据口袋染色,数据加载完成后,会自动生成与上传口袋数相同的复选框按钮,选择要染色的口袋,点击color,选中的口袋的所有氨基酸将依口袋的不同被染成不同的颜色.当上传蛋白质的残基数目较多时(例如大于300个),可滚动鼠标实现图的缩放,缩放比例范围为[0.5,2].区域II用于显示蛋白质三维结构(提供Cartoon、Spacefill、Licorice、Surface 4种显示方式)并根据计算的中心度值将其染色,将中心度值按照数值从大到小的顺序进行三等分,分别为高中心度值(显示为红色)、中中心度值(显示为绿色)以及低中心度值(显示为蓝色),用户可以在下拉列表中选择不同类型的中心度进行染色.在区域I与区域II的下方,分别显示3种中心度的区间分布图(横坐标为相应中心度的区间取值范围,纵坐标为氨基酸中心度取值在该范围内的数目)、中心度值随残基序号的折线图,不同中心度分别为横纵坐标的散点图.
3) 点击Download区域的json、jpeg文件可以打开或者下载相应文件.
利用力导向图可以更加深入和直观的了解蛋白质结构,挖掘有用的信息.

图3 页面主体功能区Fig.3 Main body of the resulting page
2.3 蛋白质残基相互作用网络的网络特征计算
以计算p38alpha c162s突变体(PDB code:1r3c)的closeness为例,上传蛋白质PDB结构文件及DogSiteScorer网站计算的口袋结构信息并提交任务[27].在结果页面,选择图3所示区域I底部的ByCentrality染色方案,右侧下拉列表中选择closeness选项,在输入框中输入0,10(表示将closeness排名前10的氨基酸染成红色),点击Color,将光标移到红色节点上方显示出残基名称、序号、centrality值及所在口袋编号,所得结果如表2.其中P_0为ATP口袋,因而一半的残基在ATP口袋,说明了ATP口袋有较强的保守性.betweenness的计算结果如表3所示.
可以看出closeness和degree近似服从正态分布,betweenness近似服从长尾分布.进一步,可下载centrality.json文件,找出betweenness最大的前10位,进行细致的分析(如表3所示).图5为p38alpha c162s突变体结构(PDB code:1r3c)的cartoon图,红色部分为betweenness值排名前10的残基,主要分布在ATP口袋周围,说明ATP口袋中的残基对蛋白质网络拓扑结构有较大的影响.值可预测蛋白质的结合位点氨基酸.
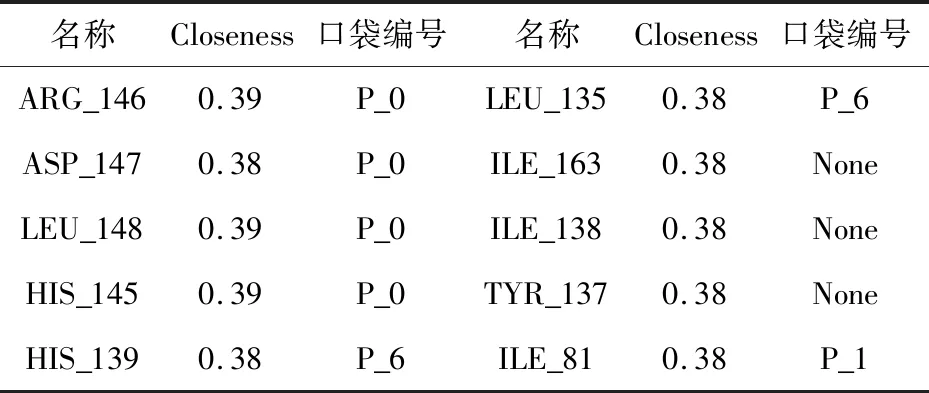
表2 1r3c中closeness值Top 10对应的氨基酸Tab.2 Top 10 amino acid by closeness of 1r3c
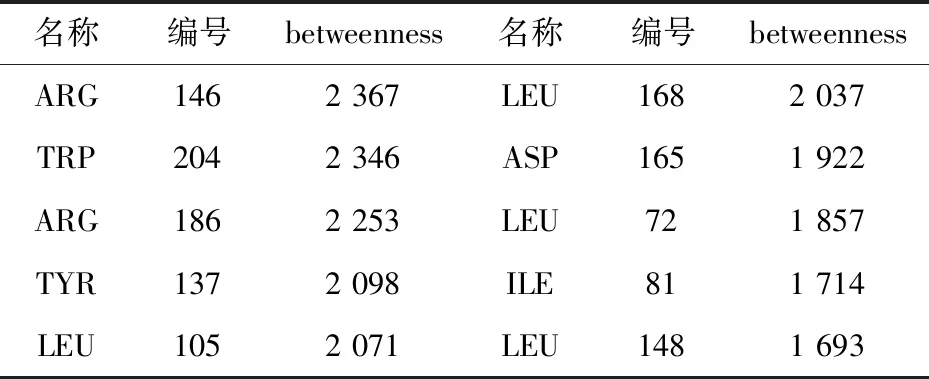
表3 1r3c中betweenness值Top 10对应的氨基酸Tab.3 Top 10 amino acid by betweenness of 1r3c
网络特征closeness centrality,betweenness centrality和degree centrality的分布规律如图4所示.
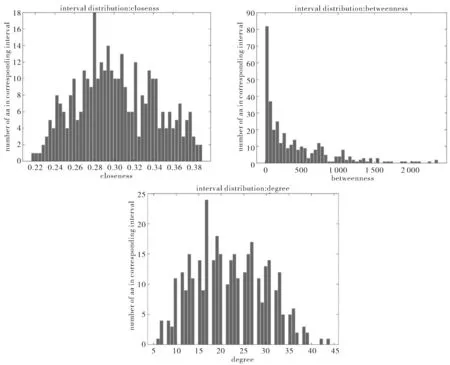
图4 centrality的分布规律Fig.4 The distribution of 3 kinds of centrality
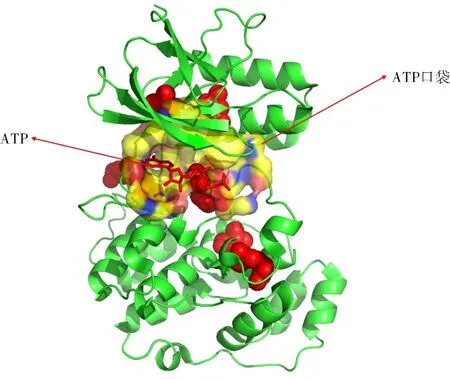
图5 p38alpha c162s突变体的结构(PDB code:1r3c)Fig.5 The cartoon presentation of 1r3c
2.4 利用closeness识别关键氨基酸
研究表明,蛋白质的活性位点,配体结合位点,进化保守残基的closeness值大多较高[28].因此,根据蛋白质残基相互作用网络中节点的closeness以枯草杆菌蛋白酶为例(PDB code:1bh6),图6为枯草杆菌蛋白酶三维结构中网络拓扑性质的closeness分布.将closeness值按照数值从大到小的顺序进行三等分,红色表示closeness值较高的残基,绿色表示closeness值中等的残基,蓝色表示closeness值较低的残基.结果表明,枯草杆菌蛋白酶表面closeness值较高的残基大多数分布在小分子的结合位点区域.

图6 枯草杆菌蛋白酶(1bh6)closeness分布图Fig.6 The distribution of closeness of subtilisin(PDB code:1bh6)
2.5 基于betweenness识别蛋白质折叠过渡态系综中的关键氨基酸[8]
研究表明,蛋白质残基相互作用网络不同于随机网络与规则网络,具有处于两者之间的小世界网络的特性,即具有较大的聚类系数和较小的平均最短路径[5].因此,利用网络节点的betweenness可以有效识别蛋白质折叠过程中过渡态系综的关键残基.Vendruscolo等[29]通过实验确定了酰磷酸酶(PDB code:1aps)折叠过程中的关键残基,具体为TYR11、PRO54、PHE94.图7为酰磷酸酶残基相互作用网络的betweenness数值分布,TYR11和PHE94的betweenness较高.

图7 1aps蛋白的betweenness值随残基序列的分布Fig.7 The distribution of betweenness of protein 1aps(pdb code:1aps )
3 总结与展望
蛋白质残基相互作用网络模型为研究蛋白质的结构与功能的关系提供了崭新的视角.网络拓扑结构中的接近中心度、中介中心度等性质,反映了单个氨基酸与蛋白质整体结构的关系[9].该方法在蛋白质稳定性,蛋白质折叠动力学,控制酶活性和变构调节,信号转导等方面取得了一系列成果.
本文利用D3.js,HTML,PHP,Javascript,MATLAB和NGL Viewer等程序模块搭建了蛋白质残基相互作用网络在线服务及可视化分析平台.用户可以利用蛋白质结构信息搭建网络结构,并计算接近中心度等网络的拓扑结构信息.长度约300个氨基酸的中等大小蛋白质,计算速度约为30 s.另外,该分析平台还可分析蛋白质的结合口袋特征,有较强的可扩展性,可添加网络最短路径,k个节点的极大完全子图(k-clique)等模块单元.结果表明,蛋白质残基相互作用网络在线服务及可视化分析平台对理解蛋白质结构,调控功能和药物设计的相关研究有重要的帮助.
