第一性原理研究B掺杂Mn4Si7的电子结构和光学性质
2020-05-15张晋敏
王 立,张晋敏,钟 义,贺 腾,王 坤,谢 泉
(贵州大学大数据与信息工程学院 贵州大学新型光电子材料与技术研究所,贵阳 550025)
1 引 言
高锰硅化合物(Higher manganese silicide,HMS)MnSi1.7是一种很好的热电材料,MnSi1.7也是一种具有高电导率、抗氧化性能好、成本低、没有污染等许多优点的环境友好型半导体材料[1,2].单晶的MnSi1.7具有烟囱梯状(Nowotny Chimney Ladder,NCL)的晶体结构[3],在块状晶体中,它有四种相结构[4]:Mn4Si7、Mn11Si19、Mn15Si26、Mn27Si47.
在近几年关于MnSi1.7的研究中,主要关注其热电性质与光电性质.在热电性质方面,刘晓虎等[5]首次采用快速凝固热压技术制备高锰硅热电材料,研究了不同Si含量对MnSi1.7热电性能的影响,电导率随Si含量的增大而下降,Seebeck系数和热导率均随Si含量的增大而上升.樊东晓等[6]采取悬浮熔炼法制备Ge取代的高锰硅试样Mn(Si1-xGex)1.733,采用甩带法制备快凝高锰硅合金粉末,快速凝固有效地降低了材料的热导率,Ge取代则提高了电导率,从而提高材料的热电性能.He等[7]分别采用热压法和电子束蒸发法制备了高锰硅(MnSi1.7)块体材料和薄膜材料,得到未掺杂块体和薄膜MnSi1.7为p型,在膜中加入铁可得到n型MnSi1.7薄膜,发现n型MnSi1.7的热电性能优于p型MnSi1.7.在光电性质方面,张民[8]首次利用第一性原理对MnSi1.7(Mn4Si7相)的电子能带结构进行了理论计算,计算结果表明Mn4Si7化合物是能隙为0.83 eV的直接带隙半导体,之后Migas等[4]计算了MnSi1.7的四种不同相的能带结构,计算表明Mn4Si7是能隙为0.77 eV的间接带隙的半导体,而Mn11Si19、Mn15Si26、Mn27Si47的行为类似于简并半导体,性质更接近金属.Gao等[9]研究了Mn4Si7的电子结构与光学性质.刘怿辉等[2]研究了硅基外延Mn4Si7薄膜电子结构与光学性质.虽然对高锰硅化合物MnSi1.7的光电性质已经做了大量的工作[10-17],但B掺杂Mn4Si7的电子结构和光学性质的理论计算尚未见报导.
采用基于密度泛函理论的(Density functional theory,DFT)第一性原理计算方法,计算未掺杂及B掺杂Mn4Si7的电子结构和光学性质,研究B掺杂对Mn4Si7的电子结构和光学性质的影响.
2 构建模型和计算方法
2.1 模型构建
Mn4Si7具有四方烟囱梯状结构,空间群为P-4c2,其晶格常数a=b=0.5525 nm,c=1.763 nm.Mn4Si7的晶胞如图1所示,Mn4Si7晶胞中有4个原胞,在晶胞中Mn原子有5 个位置,Si原子有4个位置.本文采用1×1×1的Mn4Si7超晶胞,体系共有44个原子,建立了Mn16Si28,Mn16Si27B1超晶胞模型.

图1 Mn4Si7晶胞结构 Fig.1 Cell structure of Mn4Si7
2.2 计算方法
采用第一性原理计算方法,其计算基于Material Studio软件中的CASTEP模块,CASTEP模块的计算原理是基于密度泛函理论的从头计算量子力学方法,采用总能量平面波赝势方法,由密度泛函理论中广义梯度近似(GGA)近似处理电子与电子之间的相关关联能,该方法使用广泛,并且计算电子结构较为准确.
首先采用CASTEP模块对Mn16Si28,Mn16Si27B1超晶胞进行几何结构优化,得到稳定的结构体系,再对优化后的结构体系分别计算未掺杂和B掺杂的能带结构、电子态密度和光学性质.计算选用的是平面波超软赝势来处理离子与电子的相互作用,在相互交换关联能部分,采用的是广义梯度近似中的PBE方案来处理电子与电子的相互作用,设置平面波截断能为400 eV,以及迭代过程中的收敛精度为2.0×10-6eV/atom,k点选取4×4×1.
3 结果与分析
3.1 能带结构和态密度
图2(a)为未掺杂Mn4Si7的能带结构,图2(b)为B掺杂Mn4Si7的能带结构.由图2(a)可知,未掺杂Mn4Si7是间接带隙半导体,导带底和价带顶的位置分别位于Z、G点,禁带宽度为0.786 eV,这与Migas等[4]计算的结果0.77 eV基本一致,这正是光纤通信中最重要的波段,对应光波长1.5 μm,这种波长的光在传输中具有低传输误差、低散射损耗的特点.若不考虑杂质能级的影响,在图2(b)中以可得到,B掺杂的Mn4Si7的导带底和价带顶的位置分别位于Z、G点,其禁带宽度为0.723 eV,与未掺杂的Mn4Si7相比其禁带宽度减小了.并且B掺杂以后,价带顶稍微上移,费米能级(定义为能量零点)进入了价带,这说明了B掺杂以后,此时的Mn4Si7已经是p型半导体.
为了进一步研究B掺杂对Mn4Si7电子结构变化的影响,计算了未掺杂和B掺杂Mn4Si7的态密度,图2(c)为未掺杂Mn4Si7的态密度图和各原子的分波态密度图,图2(d)为B掺杂Mn4Si7的态密度图和各原子的分波态密度图.由图2(c)可知未掺杂Mn4Si7在能量-6 eV到-4 eV这个范围,Si-3p电子轨道对态密度的贡献较大,在费米能级(定义为能量零点)附近,导带和价带主要是由Si-3p、Mn-3d态组成,其中Mn-3d电子轨道对态密度的贡献较大,在费米面附近其态密度陡然降低,表现出半导体的性质.从图2(d)可以看出,杂质B原子对价带贡献较大,对导带的贡献较小,由于B-2p的电子轨道贡献,掺杂后的态密度微微向能量负方向发生了移动.



3.2 光学性质
3.2.1复介电函数
介电函数是沟通电子跃迁微观物理过程与固体电子结构的桥梁,其反映了固体能带结构,通过介电函数能得到其它各种光谱信息.通过理论计算,得到了介电函数:
ε(ω)=ε1(ω)+iε2(ω)
(1)
公式(1)的实部ε1(ω)和虚部ε2(ω),图3(a)和图3(b)分别为未掺杂Mn4Si7和B掺杂Mn4Si7的介电函数的实部ε1(ω)和虚部ε2(ω)与入射光子能量的关系.从图3(a)中可以得到未掺杂Mn4Si7的静态介电常数为14.80,B掺杂Mn4Si7的静态介电常数则增加为17.08,未掺杂Mn4Si7在光子能量为0.63 eV处出现峰值为15.50,B掺杂Mn4Si7则在光子能量为0.51 eV处出现峰值为15.69.从图3(b)中可以得到未掺杂Mn4Si7和B掺杂Mn4Si7介电函数的虚部基本重合,在光子能量为1.42 eV处出现峰值为13.53,光子能量0到8 eV范围内,ε2的值都不为零,说明在这个光子能量区间有电子跃迁,在光子能量大于8 eV的区间,其值趋于0.
3.2.2复折射率
可以根据介电函数与复折射率的关系:
ε1=n2-k2
(2)
ε2=2nk
(3)


图3 复介电函数:(a)实部,(b)虚部.Fig.3 Dielectric functions:(a) real part,(b)imaginary part.
计算未掺杂Mn4Si7和B掺杂Mn4Si7的复折射率,计算结果如图4所示,图4(a)和图4(b)分别是折射率和消光系数与入射光子能量的关系.由图4(a)可知,Mn4Si7的折射率n0为3.847,这结果与刘等[2]计算结果基本一致,B掺杂以后,其折射率n0增加为4.137.Mn4Si7在光子能量为0.75 eV处它的折射率达到最大峰值为4.002,B掺杂Mn4Si7则在光子能量为0.69 eV处折射率达到最大峰值为4.021,峰值之后,其变化曲线基本重合,变化趋势一致,随光子能量的增大其折射率逐渐降低,在达到谷值后,其折射率又开始随光子能量的增大其折射率逐渐增加,最后折射率在0.8附近波动,综合来看,在整个能量范围内,B掺杂Mn4Si7的折射率都比未掺杂Mn4Si7的折射率大.由图4(b)可知,未掺杂Mn4Si7的消光系数在1.74 eV处达到最大峰值为2.226,B掺杂Mn4Si7的消光系数在1.73 eV处达到最大峰值为2.229,之后消光系数就随光子能量的增加而降低,在光子能量大于12 eV的区间内,消光系数趋于0.

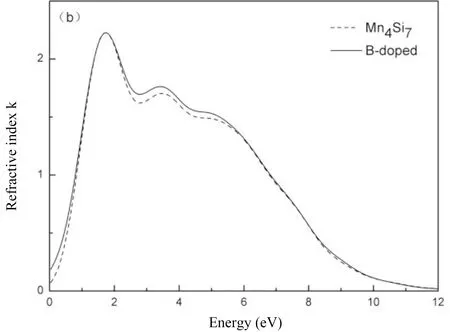
图4 复折射率:(a)折射率,(b)消光系数.Fig.4 Complex refractive index:(a)refractive index,(b)extinction coefficient.
3.2.3吸收谱和反射谱
吸收系数指的是光波在此半导体介质中传播单位距离时光强度衰减的百分比.图5(a)和图5(b)分别为未掺杂Mn4Si7和B掺杂Mn4Si7的吸收谱和反射谱.由图5(a)可知,Mn4Si7的吸收系数随着光子能量的增加逐渐增大,在光子能量为5.74 eV达到峰值1.27×105cm-1后开始逐渐变小.B掺杂Mn4Si7以后吸收系数最大值增加为1.28×105cm-1,B掺杂Mn4Si7以后吸整体的吸收系数也是比未掺杂的大.由图5(b)可知,Mn4Si7在光子能量为0时反射率为0.344,而在光子能量0到10 eV范围内,有4个峰值分别为0.422,0.349,0.520,0.536,B掺杂以后,反射率变大了.
3.2.4光电导率
光吸收使得半导体中形成非平衡载流子,而载流子浓度的增加必定使样品电导率增加,这种由于光照引起半导体电导率增加的现象称为半导体的光电导效应.图6为未掺杂Mn4Si7和B掺杂Mn4Si7的光电导率与入射光子能量的关系,如图所示,Mn4Si7的光电导率的实部在光子能量0~10 eV范围,有两个峰值,分别为2.201,2.498,B掺杂Mn4Si7的两个峰值分别为2.279,2.513,B掺杂引起Mn4Si7的光电导率增加.
4 结 论
采用基于密度泛函理论的第一性原理计算方法,先计算未掺杂Mn4Si7的电子结构和光学性质,再计算B掺杂Mn4Si7的电子结构和光学性质并与未掺杂Mn4Si7的计算结果进行对比.计算结果表明,未掺杂Mn4Si7是间接带隙半导体,其禁带宽度为0.786 eV,B掺杂以后其禁带宽度下降为0.723 eV;未掺杂Mn4Si7在费米能级附近,导带和价带主要是由Si-3p、Mn-3d态组成,其中Mn-3d电子轨道对态密度的贡献较大,杂质B原子对价带贡献较大,对导带的贡献较小,由于B-2p的电子轨道贡献,掺杂后的态密度微微向能量负方向发生了移动,B掺杂Mn4Si7是p型半导体材料;未掺杂Mn4Si7的吸收系数在近红外区达到105cm-1,Mn4Si7的静态介电常数为为14.80,B掺杂以后静态介电常数增加为17.08,Mn4Si7的折射率n0为3.847,B掺杂以后,其折射率n0增加为4.137,总之B掺杂引起Mn4Si7的折射率、吸收系数、反射系数及光电导率增加.

图5 (a)吸收谱;(b)反射谱.
Fig.5 (a) Absorption spectrum;(b)Reflection spectrum.

