三氯苯酚在TiO2 (101)面吸附特征的理论研究
2020-05-15王丹阳刘柳斜李来才
罗 惠, 王丹阳, 刘柳斜, 李来才
(四川师范大学 化学与材料科学学院, 成都610066)
1 引 言
氯代酚类化合物大多具有致癌性、致突变和致畸变作用以及高毒性、持久性和生物蓄积性等特点[1, 2].三氯苯酚既是合成燃料、防腐剂、杀菌剂咪鲜胺、除草剂草枯醚等的中间体,也是降解咪鲜胺、草枯醚等物质的产物之一,是最有害的环境污染物之一[3-5].美国及欧盟将其列入“优先控制污染物”名单,我国也将其列入了68种水环境优先检测污染物黑名单当中[6].目前关于如何有效处理三氯苯酚的技术研究已经引起了人们的广泛关注.目前的处理方法集中在:物理、化学、生物以及各种方法的结合.其中物理法最常见的为吸附法,利用吸附材料对环境中的三氯苯酚进行吸附处理[7],但从根本上来说只能达到物理处理效果,并不能消除三氯苯酚的本体.生物法可以利用微生物的代谢作用从根本上消除污染[8],但是三氯苯酚本身具有毒性,培育带有针对代谢功能的微生物具有相当大的难度,微生物本身对水资源以及土壤的污染也应该另做研究.因此,利用光催化降解三氯苯酚的化学降解法就成为了目前的研究热点.其中,TiO2光催化是一种极具潜力的高级氧化技术,它的优点是反应条件温和,稳定性高,操作简单,价格低廉且无二次污染[9],在处理难降解有机污染物方面具有很大的应用潜力.Hee-Chan等研究了TiO2的表面改性、焙烧温度和初始pH值等因素对三氯苯酚的光催化活性的影响[10].Tanaka等研究了在302-405 nm波长范围内的高压水银灯照射时,pH值对TiO2悬浮液光催化吸附和分解三氯苯酚的影响[11].目前对锐钛矿型TiO2催化降解三氯苯酚的吸附位点和反应机理的研究还无报道.本文利用密度泛函理论,对三氯苯酚在锐钛矿型TiO2(101)晶面的吸附特征进行了研究,对于了解三氯苯酚吸附在TiO2表面上的结构变化有重要意义.希望能为后续降解处理三氯苯酚提供有价值的信息.
2 计算方法
本章的所有研究采用的计算模块是基于第一性原理[12, 13]的MS软件中的CASTEP,平面波截断能采用Rc=260 eV,迭代过程中的收敛精度设置为2.0×10-6eV.采用超软赝势来描述离子实与价电子之间的相互作用,忽略内层电子效应.电子之间的交换关联能采用广义梯度近似GGA加PBE进行处理[14].除此之外的其他数据均采用程序默认值.在结构优化过程当中,三氯苯酚的总体能量最小值由CASTEP模块中的BFGS方法[15]寻找.参考其他研究人员利用第一性原理对TiO2(101)晶面电子结构的研究[16-19],构建了TiO2(101)晶面模型.TiO2(101)面采用的参数为:a=10.8858 Å,b=11.328 Å,c=30.000 Å;α=β=90°,γ=110°.顶层TiO2表面正上方留有约10 Å的真空层,用于放置吸附原子,且每个TiO2晶胞结构中均含有36个Ti和72个O原子.吸附能Ead采用如下公式进行计算:
Ead=Ecomplex-ETiO2-ETCP
Ecomplex是指三氯苯酚分子吸附在TiO2(101)晶面时体系的总能量,ETiO2是TiO2(101)晶面的能量,ETCP是三氯苯酚的能量.
3 结果与讨论
3.1 TiO2(101)面吸附三氯苯酚的吸附能及结构变化分析
三氯苯酚有四个可降解的位点,分别是羟基邻位的氯原子Cl、羟基对位的氯原子Cl(2)、羟基上的氢原子H、羟基间位上的氢原子H(2).我们选择了TiO2(101)面上的四个吸附位点,分别是二配位的氧原子O(2)、三配位的氧原子O(3)以及五配位的钛原子Ti(5)、六配位的钛原子Ti(6).分析过程中使用“-”表示吸附结合.由此,三氯苯酚在锐钛矿TiO2(101)晶面存在着16种不同的吸附方式,其吸附构型见图1.为了找出稳定的吸附构型,我们计算了每种吸附模型的吸附能,如表1所示.根据表1的数据可知,O(2)-Cl(2)、O(3)-H、Ti(5)-H和Ti(6)-H(2)此四种吸附构型的吸附能较大,为该研究获得的稳定吸附模型.为了进一步探讨三氯苯酚是否具有断键降解的可能性,我们选取了这四种稳定模型的优化结构进行具体的讨论.

图1 三氯苯酚在TiO2 (101)表面的吸附结构Fig.1 Adsorption structures of TCP adsorbed on TiO2 (101) surface
稳定吸附模型的优化图如图2所示.O(3)-H构型中吸附位相邻C-C键长分别由1.307 Å、1.308 Å增加到1.394 Å、1.403 Å,分别增加了0.087 Å、0.095 Å,C-Cl键长分别由1.620 Å、1.634 Å增加到1.715 Å、1.735Å,分别增加了0.095 Å、0.101 Å.在O(2)-Cl(2)构型中,吸附位相邻的C-C键长分别由1.295 Å、1.296 Å增加到1.392 Å、1.393 Å,均增加了0.097 Å.Ti(5)-H结构中吸附位相邻C-C键长分别由1.307 Å 、1.308 Å增加到1.403 Å 、1.407 Å,分别增加了0.096 Å、0.099 Å,C-Cl键长分别由1.620 Å、1.634 Å增加到1.718 Å、1.725 Å,分别增加了0.098 Å和0.091 Å.Ti(6)-H(2)吸附结构中吸附位相邻C-C的键长分别由1.293 Å、1.295 Å 增加到1.387 Å、1.396 Å,分别增加了0.094 Å、0.101 Å, C-Cl键长分别由1.632 Å、1.634 Å增加到1.734 Å、1.740 Å,分别增加了0.102 Å、0.106 Å.O(3)-H构型中,吸附位相邻的∠O-C-C分别由119.07°、123.41°变化为118.48°、113.70°,键角有明显变化.如图,其他三种构型的键角也有相应的变化.

表1 三氯苯酚在TiO2 (101)表面的吸附能
根据表1吸附能和图2键长变化的分析,发现O(3)-H构型的吸附能最大,吸附最稳定,吸附位C- Cl键长和C-C键长均有增加,键角也有明显的变化,C-Cl键和C-C键得到了活化,使得相应位置键断裂的可能性增加.由此可知:O(3)-H构型为最稳定的吸附构型.
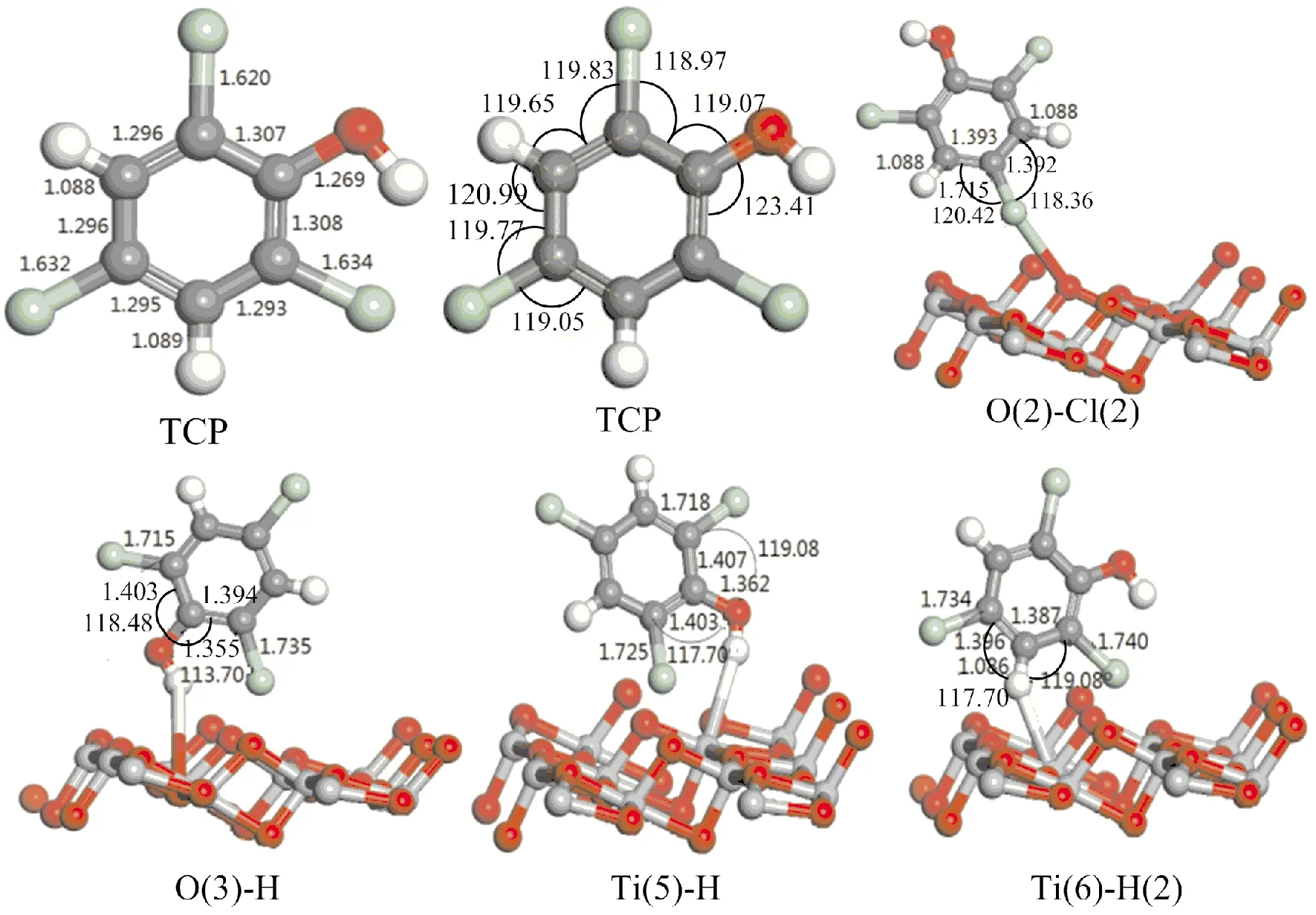
图2 三氯苯酚在TiO2 (101)表面稳定吸附结构的键长(Å)和键角(°)Fig.2 Bond lengths (Å) and bond Angles (°) of stable adsorption structures of TCP adsorbed on TiO2 (101) surface
3.2 二氧化钛吸附三氯苯酚的电子结构分析
为了进一步了解三氯苯酚与TiO2(101)晶面之间的电子相互作用,我们列出了这四种稳定吸附构型的态密度图、分态密度图以及电荷密度图.分别见图3、图4.

图3 三氯苯酚在TiO2 (101)表面稳定吸附的态密度图和分态密度图Fig.3 DOSs and PDOSs of stable adsorption structures of TCP adsorbed on TiO2(101) surface

图4 三氯苯酚在TiO2 (101)表面稳定吸附结构的电荷密度图Fig.4 The electron density maps of stable adsorption structures of TCP adsorbed on TiO2 (101) surface
如图3所示, 对比Cl(2)原子与TiO2晶胞上O(2)配位吸附, H(2)原子与TiO2晶胞上Ti(6)配位吸附及H原子与TiO2晶胞上O(3)、Ti(5)配位吸附的总态密度图与未吸附时TiO2的总态密度图,可以发现这四种吸附构型吸附后费米能级附近的电子数有明显减少.对比吸附前后的分态密度图得知,这是因为被吸附的Cl原子和H原子与TiO2晶胞上的吸附位原子发生了P电子峰重叠,说明此四种吸附方式可以产生一定程度的电子相互作用,为化学吸附.
图4给出了这四种稳定吸附构型的电子密度图.从图中可以清楚地看到三氯苯酚与TiO2(101)面之间的成键情况.O(3)-H、O(2)-Cl(2)、Ti(5)-H和Ti(6)-H(2)四种吸附构型中三氯苯酚与TiO2(101)晶面之间存在电子云的重叠,产生了一定程度的电子相互作用,有新的化学键生成,符合化学吸附的特征.此结论与态密度及分态密度的分析结论一致.
4 结 论
本文采用基于密度泛函理论的第一性原理计算方法对比研究了具有代表性的TiO2(101)晶面吸附三氯苯酚的吸附特征和本质.通过对吸附构型的吸附能、态密度及电荷密度的分析,可以看出三氯苯酚中的H原子和Cl原子能被TiO2稳定吸附,存在四种稳定吸附构型且具有化学吸附的特征.而三氯苯酚羟基上的H原子被吸附在TiO2(101)面的三配位的O原子上时,吸附能最大,是最佳吸附构型,吸附后三氯苯酚与TiO2(101)面产生了电子相互作用,使部分键得到了活化.由此说明利用锐钛矿TiO2催化降解三氯苯酚具有理论可行性.
