N2在单原子催化剂Ir/MoS2表面上吸附的第一性原理研究
2020-05-15肖香珍蔡颖莹张建伟
肖香珍, 蔡颖莹, 张建伟
(1.河南科技学院 实验管理中心, 新乡 453003; 2.河南工学院 材料科学与工程学院, 新乡 453003)
1 引 言
对于高负载量的金属催化剂,无论是在实验研究或理论计算上已有相当成熟的研究报道[1-4]但在催化反应中传统负载型金属催化剂的金属利用效率远远低于理想水平[5-7],只有极少数金属活性组分起催化作用,而目前单原子催化剂实现了原子利用率的最大化,且单原子催化剂有着高活性、高选择性和高稳定性的突出优势.单原子催化的研究取得了很多突破性的研究,如张涛课题组[8]成功制备出单原子Pt1/FeOx催化剂,在CO氧化反应中表现出很高的催化活性和稳定性;另外将Pt[9-11]、Ir[12, 13]、Au[14]等贵金属吸附在不同铁氧化物上,对水煤气变换、CO氧化、NO还原等反应都获得了很好的催化性能.DFT理论计算也是催化实验研究的重要辅助手段,通过理论计算模拟出近似真实的催化剂,进而揭示单原子催化剂的催化反应机理,为研究及开发新型的单原子催化剂奠定基础.目前研究者们已经开展了大量的DFT 理论计算,如:单原子可能的吸附位点包括表面吸附、取代载体嵌入等.不管单原子是吸附在载体表面还是嵌入到载体结构中,单原子催化剂都需要合适的载体,通常选用的载体是金属及其金属氧化物,目前二维材料石墨烯[15, 16]、六方氮化硼( h-BN)[17]等作为载体应用前景较大.例如:Guo 等[18]通过理论计算考察了不同单原子M(Ag,Au,Pt,Rh,Pd,Fe,Co和Ir ) 负载在具有B空缺的六方氮化硼(h-BN)上的CO 氧化机理.
地球上氮的循环利用其中一个重要的角色是合成氨,传统的合成氨需要高温高压,对操作条件及反映设备要求也高,相比之下,光催化氮还原制氨和电催化氨合成有着节能和环境友好的优势,因此越来越受到关注[19].那么,研究氮气还原成氨的反应过程与机理,首先要了解N2在催化剂上的吸附性能.本论文是在之前研究过渡金属Ir催化剂表面性质基础上[20, 21]以单层MoS2作为载体,单原子Ir附着在MoS2载体上形成单原子催化剂Ir1/ MoS2,采用密度泛函理论(DFT)的第一性原理,计算分析了N2在单原子催化剂Ir1/ MoS2吸附的偏好位置以及吸附的电子结构.
2 计算模型和方法
MoS2是一种天然的新型半导体材料[22],晶体结构类似于石墨烯,属于六方晶系且具有二维层状结构,每一层都是三明治式S-Mo-S的结构,即Mo 原子在中间,上下分别由共价键连接着S原子,三个原子层中的原子都按类似石墨烯的平面六角阵列方式排列.
本文采用Slab模型来模拟单原子催化剂表面,所用的是3×3 单层MoS2超胞,由9个Mo原子和18个S 原子组成,所建超胞模型的俯视图和侧视图如图1所示,结构优化后本征单层MoS2的晶胞参数为0.319 nm,与实验值0.316 nm[23]接近,证明计算结果是可信的.所有计算均采用DFT方法[24-26],本文计算采用了基于密度泛函理论平面波赝势方法的VASP[27, 28]软件包,单电子波函数采用平面波基组展开,截断能为500 eV,交换相关能采GGA-PW91[29],离子芯势采用PAW方法描述[30],不可约布里渊区积分采用Monkhorst-Pack方案[31],K点网格密度取为5×5×1.在构型优化过程中,吸附物Ir和N2放在表面一侧,顶层S原子随吸附物驰预,其余Mo和S原子固定,为了避免MoS2在z轴方向的影响,真空层厚度取为15 Å.所有优化得到的最优构型都通过频率分析加以验证,即局域极小点对应全部实频.

图1 单层MoS2超胞的俯视图和侧视图及单原子催化剂Ir1/MoS2,黄色为S原子,紫色为Mo原子,深黄色为Ir原子.Fig.1 Top view and side view of single layer MoS2 supercell and single-atom catalysts Ir1/MoS2.Yellow is S atom, purple is Mo atom, and dark yellow is Ir atom.
3 结果与讨论
3.1 过渡金属吸附在单层MoS2载体上的最优位置
为了明晰N2吸附在单原子催化剂Ir1/MoS2上的本质,本文首先通过计算和比较不同的吸附构型的总能来确定体系Ir1/MoS2的稳定性和形态.根据MoS2的结构,我们考虑了Ir原子分别附着在四重空位(fcc位)和三重空位(hcp位)两个典型的吸附位点.Ir原子附着在MoS2表面的四重空位(fcc位)的总能量为E=-208.91295296 eV,hcp位为E=-209.91335529 eV,频率分析也得到hcp位对应全部实频,无虚频,显示hcp位相对稳定,其结构俯视图示于图1(c).
3.2 N2在体系Ir1/MoS2上吸附的几何结构
吸附能的定义是指吸附前后各物质总能量的变化, 其符号和大小可以表示发生吸附的可能性与吸附的程度[32].本文结合前面的吸附模型,吸附能均依下式计算:
Eads=EN2+EIr/MoS2-EN2/Ir/MoS2
(1)
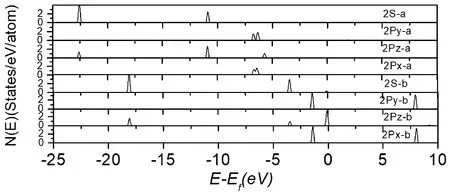
其中,EN2、EIr1/MoS2、EN2/Ir1/MoS2分别表示吸附分子N2、Ir1/MoS2催化剂表面以及N2吸附体系Ir1/MoS2的总能量.表1给出了N2在Ir1/MoS2上吸附的吸附能及几何结构.如果计算后Eads>0,则表明粒子被吸附, 吸附能愈大, 则吸附愈稳定;反之,Eads 氮气在Ir1/MoS2上的吸附类似于铁/Mxene、Co/Mxene、Ru/Mxene和Rh/Mxene上的吸附[33], N2吸附更倾向于通过N端吸附在单原子金属Ir的顶位上,几种吸附构型如图2所示.表1中列出了几种构型的N-N键长、N-Ir键长以及对应的吸附能,说明在此列出的几种构型中N2分子均为化学吸附,具有较高的吸附能,依次为1.57、1.53、1.51、1.43 eV,见表1,相对于纯的金属Ir(100)表面N2在顶位的吸附能0.78 eV[34],此吸附更加稳定,因此表明单原子催化剂Ir1/MoS2有一种强烈的氮缺乏症,而且与金属Ir作为催化剂比较,大大减少了贵金属的使用量,降低了成本. 图2 N2在Ir1/MoS2表面上的几种吸附构型图Fig.2 Several adsorption configurations of N2 on Ir1/MoS2 surface 这四种吸附构型中的N-N键长度分别为1.14 Å、1.14 Å,1.13 Å、1.14 Å, 略长于气相N2(1.11Å)中的键长,类似于Tm/Bn催化剂中的N-N键长约为1.12 Å[35],说明N2在单原子催化剂Ir1/MoS2的吸附对N-N 键未有扰动. 表1 N2在Ir1/MoS2表面上的吸附优势结构及吸附能 Table 1 The geometries and energies for N2adsorbed on Ir1/MoS2 (a)(b)(c)(d)Eads(ev)1.5701.5351.5191.431dN-N(Å)1.141.141.131.14dIr-N(Å)1.921.921.921.91 Eads,dIr-N,dN-Nare the adsorption energy, the distance of N atom to the metal Ir atom, the N-N bond length. 电荷密度差分图可以反映N2吸附前后电荷密度的重新分布情况.我们采用下式计算电荷密度差: Δρ=ρ(N2/Ir1/MoS2)-ρ(Ir1/MoS2)-ρ(N2) (2) 其中,Δρ表示体系的电荷密度差,ρ(N2/Ir1/MoS2)、ρ(Ir1/MoS2)和ρ(N2)分别表示吸附体系、Ir1/MoS2单原子催化剂表面以及吸附物的电荷密度. 对图3进行分析,发现N原子与N原子中间以及Ir原子下方出现电荷的降低,这说明在Z方向上N2和金属Ir的部分轨道能量发生了转移,且转移到了N原子与吸附点Ir原子的中间,这样就出现N原子与吸附点Ir原子中间区域电荷明显积聚,这就是N2在Ir1/MoS2体系中金属原子Ir上吸附成键的主要特征. 图3 电荷密度差分图,深灰色为电荷密度降低的区域,浅灰色为电荷密度增加的区域.Fig.3 Charge density difference map.Dark gray is the area where charge density decreases and light gray is the area where charge density increases. 为了进一步弄清楚N2的各轨道与体系Ir1/MoS2上的金属Ir的各轨道相互作用,我们计算了N2/Ir1/MoS2体系的分波态密度,计算结果示于图4,图5. 我们计算了N2吸附前后各个轨道的分布情况,图4是N2吸附前后各轨道的DOS图,带有字母a的表示N2吸附后的2S、2Py、2Pz、2Px的DOS图,字母b的表示气相的几个轨道DOS图,见图4.通过对图4分析,可以看到N2的2Py、2Px和2Pz均参与了作用,吸附后,2Py、2Px轨道峰明显变宽,且均明显向低能量方向发生移动,特别是2Pz轨道,向低能方向移了近4.0 eV,作用最强,从上面图4的电荷密度差分图上也可以看到主要是在Z方向上与底物金属原子Ir相互混合,使得N2能够在此表面上吸附. Note: The DOS diagrams of 2S, 2Py, 2Pz and 2Px orbitals with a representation projected to the N atoms after adsorption, and the DOS diagrams of the orbitals with b representation projected to the corresponding gaseous N atom.图4 N2/Ir1/MoS2体系的态密度,将态密度投影至N2吸附前后相应N原子的2S、2Py、2Pz和2Px轨道,费米能级为零点.Fig.4 The density of states of N2/Ir1/MoS2 system is projected to the 2S, 2Py, 2Pz and 2Px orbitals of corresponding N atoms before and after N2 adsorption.The Fermi level is zero. 图5左边图(a)为未吸附N2的金属Ir原子各轨道的投影态密度,图5右边图(b)为吸附N2后的金属Ir原子各轨道的投影态密度,对两图进行比较,可以看出吸附后Ir的5dz2轨道能量向低能方向位移最为明显,另外5dxz、5dyz、6Pz各轨道峰与未吸附前相比较,均有不同程度的峰位变宽或向低能移动.综合分析电子结构图,N2的吸附成因是吸附N原子的部分轨道与底物金属Ir的5dz2、5dxz、5dyz、6Pz有着不同程度的相互作用,其中贡献最大的是Z方向上的各轨道,即吸附N原子的2Pz轨道与金属Ir的5dz2、6Pz轨道存在混合作用,这样才出现N2吸附在顶位较稳定. 图5 N2/Ir1/MoS2体系的态密度,将态密度投影至金属Ir的6S、 6Py、6Pz、6Px 、5dxy 、5dyz 、5dZ2 、5dxz 、5dx2-y2 轨道,费米能级为零点.Fig.5 The density of states of N2/Ir1/MoS2 system is projected to the 6S, 6Py, 6Pz, 6Px, 5dxy, 5dyz, 5dZ2, 5dxz, 5dx2-y2 orbits of metal Ir.The Fermi level is zero. 采用DFT方法计算了N2在单原子催化剂Ir1/MoS2表面上吸附的几何结构与电子结构,N2偏好垂直吸附在Ir原子的顶位,吸附能为1.57 eV,吸附能较高,体现出单原子催化良好的催化性能.吸附N原子的2Py、2Pz、2Px轨道与 Ir原子的5dz2、5dxz、5dyz、6Pz轨道均存在不同程度的混合,特别是在Z方向上N的原子2Pz与Ir原子的5dz2、6Pz轨道相互作用是N2稳定吸附于表面的原因,Ir原子的其他轨道没有直接参与吸附成键,但其电荷对N2的吸附有促进作用.对于进一步开发单原子催化剂Ir1/MoS2潜在的性能和成本优势,更要注重理论计算与实验研究相结合.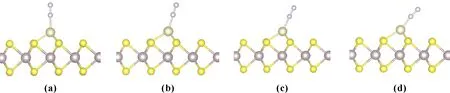
3.3 N2在体系Ir/MoS2上吸附的电子结构


4 结 论
