大体积进样气相色谱串联质谱法测定饮用水中29种半挥发性有机物
2020-05-14陈红波夏慧丽方如意
陈红波 - 夏慧丽 - 方如意 -
(台州市食品检验检测中心,浙江 台州 318000)
水资源是人类生产生活不可或缺的重要资源之一。为更好地保护生态环境和保障人民生活健康,国家对水资源的质量要求也在不断提高。GB 5749—2006《生活饮用水卫生标准》和GB 3838—2012《地表水环境质量标准》对水中多达100余种指标进行了严格的浓度限定,其中半挥发性有机污染物(SVOCs)是主要的监测目标物之一,两个标准限定的SVOCs主要是氯苯类、苯酚类、多环芳烃、硝基苯类、有机氯农药、有机磷农药等类型。这些物质多有致癌、致畸和致突变性,通过饮用水和食品等进入人体内,将对人体健康造成严重危害[1-2]。
饮用水中SVOCs的分析测定主要存在基质复杂多样、异构体多和组分复杂等问题,目前采用的分析方法多为气相色谱法[3-5]、气相色谱/质谱法等[6-8],气相色谱法仅依靠保留时间作为定性依据,容易造成假阳性或者假阴性结果,且通常需要多次进样才能完成对一个样品中多种SVOCs的分析;气相色谱/质谱法近年来得到越来越多的使用,虽然能够实现一次进样同时分析多种SVOCs,但单四级杆气相色谱质谱联用仪对某些组分的检测灵敏度不够,容易因过多基质干扰而造成检出限偏高,难以满足多组分痕量分析的需要。常用预处理方法有液/液萃取[9-10]、固相萃取[11-14]和固相微萃取[15-16]等,固相萃取技术由于所用萃取填料对各特性化合物的吸附性能不同,较难同时准确测定不同类别的SVOCs,且不同分析对象往往需要采用不同类型的萃取剂;固相微萃取受萃取纤维种类、厚度、萃取方式等诸多影响,不同分析对象需选择不同的支撑体和涂层,且成本高,稳定性较差。相对而言,液/液萃取前处理法具有萃取范围广、萃取率高、操作简便等突出优点,较适用于含有多种类别SVOCs的提取[17]。为满足饮用水中各SVOCs的极低限量要求,在前处理过程中,通常需要先对水样进行浓缩富集,费时费力,且容易带来较大的基质干扰。试验拟采用液液萃取—气相色谱—三重四级杆串联质谱仪大体积进样方式对采集的水样进行检测分析,以期简化前处理步骤,提高工作效率,实现一次进样同时分析饮用水中的多种SVOCs。
1 材料与方法
1.1 仪器与试剂
1.1.1 仪器
串联三重四级杆气相质谱联用仪:Agilent 7890B+7000D型,配多模式进样口,美国安捷伦科技公司;
超纯水机:Milli-Q Advantage,美国默克密理博公司。
1.1.2 试剂
甲基叔丁基醚:色谱纯,美国天地有限公司;
无水硫酸钠、氯化铵、磷酸氢二钠、磷酸二氢钾:分析纯,江苏强盛功能化学股份有限公司。
1.1.3 标准物质
29种半挥发性有机物标准品[1,3,5-三氯苯、1,2,4-三氯苯、1,2,3-三氯苯、六氯苯、2,4,6-三氯苯酚、五氯酚、α-六六六、β-六六六、γ-六六六、δ-六六六、莠去津、呋喃丹、乐果、七氯、百菌清、甲基对硫磷,甲醇介质;马拉硫磷、毒死蜱、对硫磷、敌敌畏,丙酮介质;灭草松、溴氰菊酯、苯并(α)芘、O,P’-DDE、P,P’-DDE、P,P’-DDD、O,P’-DDD、O,P’-DD、P,P’-DDT,正己烷介质]:100 μg/mL,上海安谱实验科技股份有限公司;
3种内标标准品(苊-d10、菲-d10、屈-d12,丙酮介质):500 μg/mL,上海安谱实验科技股份有限公司。
1.2 试验方法
1.2.1 水样预处理 取水样30 mL至50 mL离心管中,加入磷酸缓冲盐(配比:磷酸氢二钠0.5 g、磷酸二氢钾49.5 g、氯化铵0.3 g)0.5 g,调节水样的pH值为4.8~5.5,加入3 mL甲基叔丁基醚(MTBE)和12 g无水硫酸钠,振荡5 min进行液液萃取,取1 mL上层有机相至进样小瓶中,加入内标物上机测定。
1.2.2 标准溶液配制 各标准溶液用丙酮逐级稀释至各不同浓度(5.00,2.00,1.00,0.25,0.10 μg/mL)的标准溶液。
在空白水样中加入不同浓度的目标物,逐一进行前处理,得到萃取液的标准溶液,取1.0 mL萃取标准溶液加入内标物后上机测试。
各内标溶液(0.5 mg/mL)用甲基叔丁基醚逐级稀释至5 μg/mL,取1 mL最终萃取液加入内标物10 μL,萃取液中各内标物浓度50 ng/mL。
1.2.3 色谱和质谱条件
(1)色谱条件:DB-35MS UI快速分析柱 20 m×180 μm×0.18 μm(J&W121-3822UI);高纯氦气;进样量10 μL;进样口PTV模式;进样口50 ℃保持0.11 min,以900 ℃/min升至325 ℃;柱流速0.8 mL/min恒流;柱温50 ℃保持1.5 min,然后以50 ℃/min升温至150 ℃,再以25 ℃/min升温至310 ℃保持5 min,总运行时间14.9 min,使用保留时间锁定功能,将内标氘代菲保留时间锁定至7.55 min。
(2)质谱条件:扫描方式MRM模式;EI源电离,电压70 eV;离子源温度280 ℃;四级杆温度(Q1和Q2)150 ℃;传输线温度300 ℃。29种半挥发性有机物与内标物的MRM条件如表1所示。
2 结果与讨论
2.1 水样前处理用试剂的选择
水样前处理选用磷酸盐缓冲液调节水样的pH,原因是磷酸缓冲液可阻止水样中碱性物质对卤乙腈类等化合物的分解,并可除去水样中自由态的氯,保证样品pH值的一致性[18];选用甲基叔丁基醚作为萃取溶剂,是因为甲基叔丁基醚微溶于水,对多数SVOCs具有良好的溶解性,且成本较低[19];在前处理过程中加入Na2SO4有助于提高水相的离子强度,降低甲基叔丁基醚在水相中的溶解,无乳化现象,且分层较好,提高了萃取效率,同时避免水相和有机相间的乳化现象。
2.2 试验条件选择
2.2.1 溶剂放空模式大体积进样 采用10 μL大体积进样,可以大大简化前处理过程,不需要经过浓缩仍可以得到较低的检出限,通过增大进样量降低方法的检出限和对样品使用量的要求。在冷却的进样口衬管中用高载气流速蒸发溶剂,可使半挥发性化合物在衬管中浓缩,消除了繁复的手动浓缩步骤,降低了溶剂消耗量,减少了环境污染,可显著提高实验室的分析通量、提高检测灵敏度、提高工作效率、降低操作成本。
采用溶剂放空模式可以减少峰拖尾,在PTV模式下,进样量大容易使衬管变脏,引起峰拖尾,为消除这种影响,试验采用900 ℃/min快速升温至325 ℃,使分析物瞬间汽化,减少分解。进样结束后,打开分流阀,可以将衬管内高沸点、难挥发的杂质从分流阀排出,这样既清洁了进样口,又避免了杂质进入色谱柱,有效地减少了峰拖尾和待测物的吸附及分解[20]。
2.2.2 MRM多反应离子监测模式 MRM多反应离子监测即母离子被选定,碰撞后,从形成的子离子中再次选定进行监测。经过两次筛选,MRM模式比SIM模式具有更强的降噪性能,因此采用MRM多反应离子监测模式可以极大地去除基质带来的干扰。

表1 29种半挥发性有机物与内标物的MRM条件
2.3 标准溶液色谱图分析
饮用水中29种SVOCs的总离子色谱图与部分化合物MRM提取离子图分别见图1、2。
由图1可见,样品的总运行时间14.9 min,实现了一次进样同时分析29种化合物,大大提高了分析效率。由图2可见,在MRM多反应离子监测模式下,各化合物经过定量离子对与定性离子对的提取,均实现了完美分离,且各化合物均无基质干扰,响应良好,能够很好的实现饮用水中29种SVOCs的定性与定量分析。

图1 29种化合物的总离子流图

图2 部分化合物的MRM提取离子图
2.4 方法线性及检出限
在空白水样中加入不同浓度的SVOCs混合标准溶液,配制成梯度浓度的标准水溶液,然后分别进行样品前处理,取其萃取有机相加入内标后进样,绘制标准曲线。各化合物的线性范围、线性相关系数和检出限见表2。
由2表可知,在标准系列浓度范围内,各组分方法线性关系良好,r值均在0.994 0以上,呈现良好的线性关系。除苯并(α)芘外,各SVOCs的检出限均远低于限量指标,满足检测需求。
2.5 精密度与回收率
在水样中加入标准溶液,分别进行低、中、高3种不同浓度的加标试验,每个浓度进行6次平行测定,计算平均回收率和相对标准偏差。以平均回收率表示方法的准确度,以相对标准偏差表示方法的精密度。29种SVOCs的平均回收率范围为61.66%~108.62%,相对标准偏差范围为1.53%~8.71%,满足GB/T 27404—2008方法学要求。各组分回收率及相对标准偏差见表3。
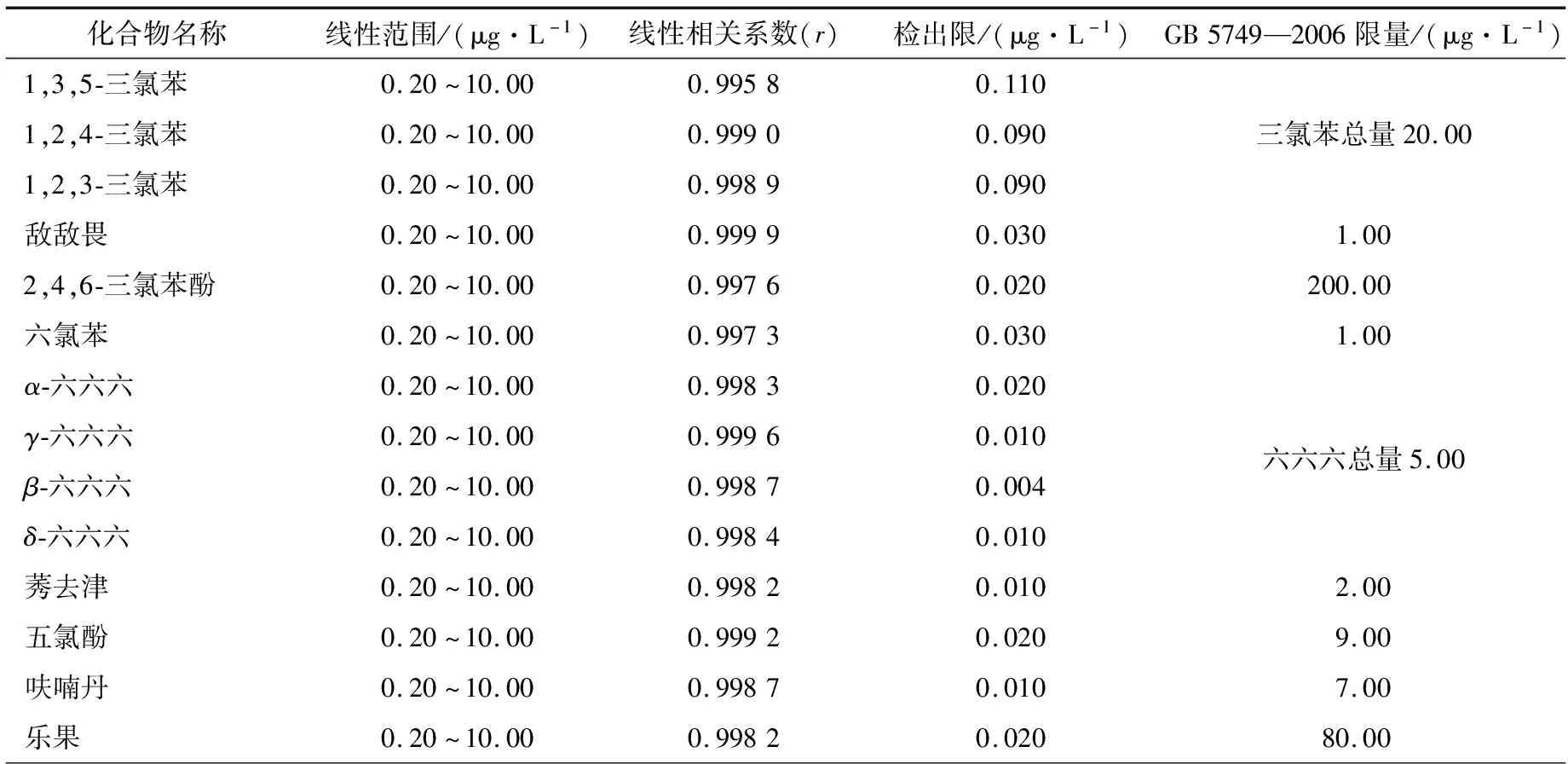
表2 29种SVOCs的线性范围、线性相关系数和检出限†
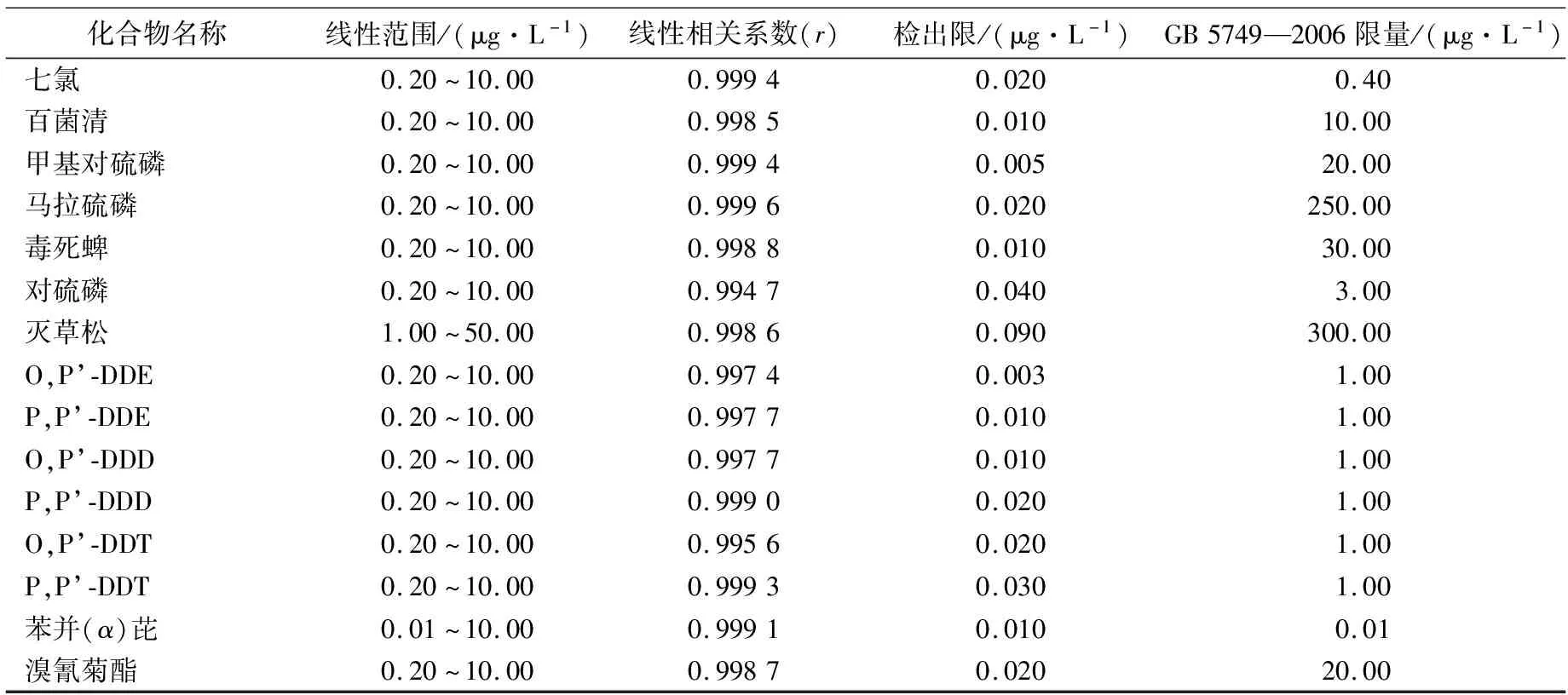
续表2
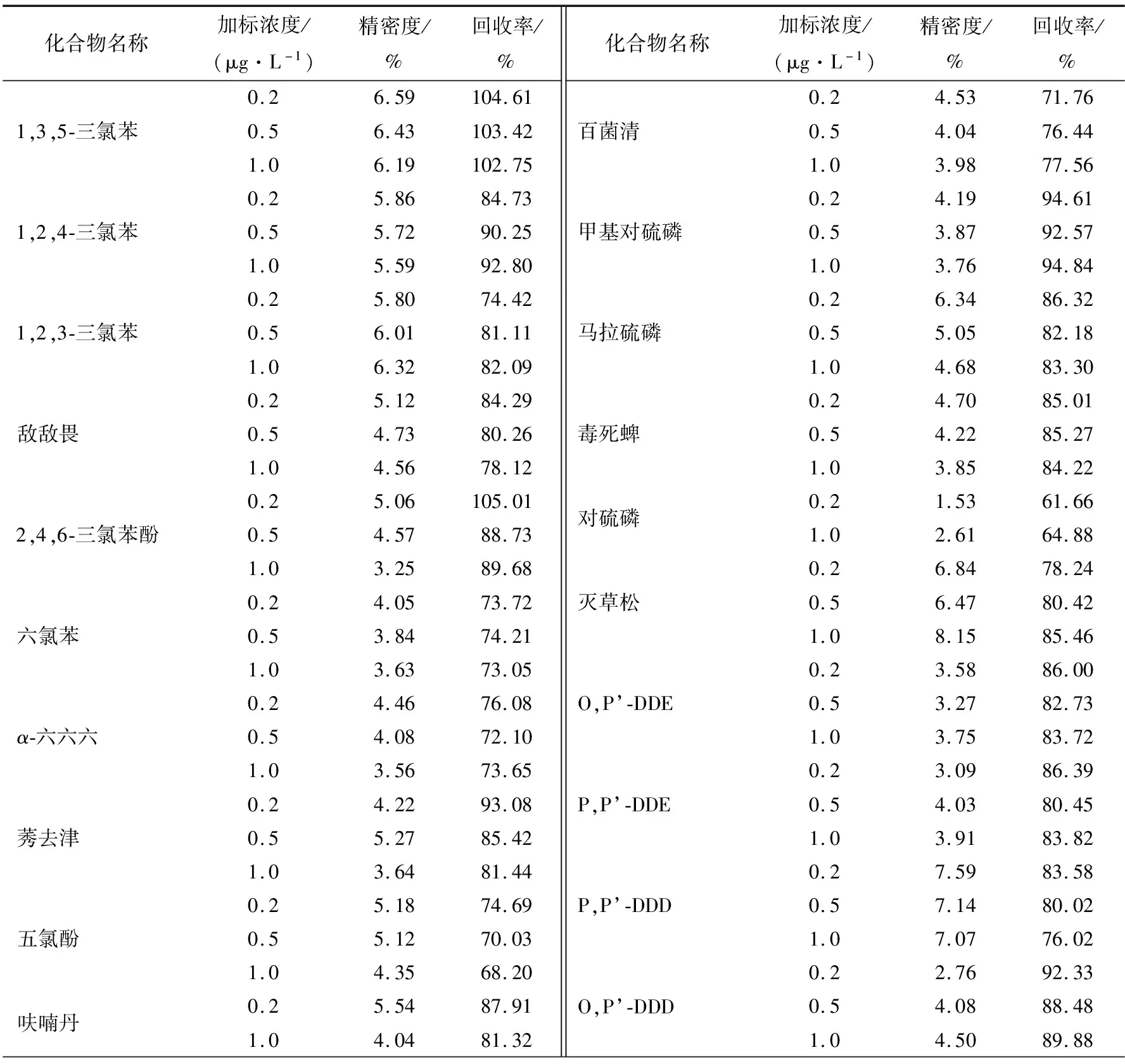
表3 各化合物的精密度与回收率
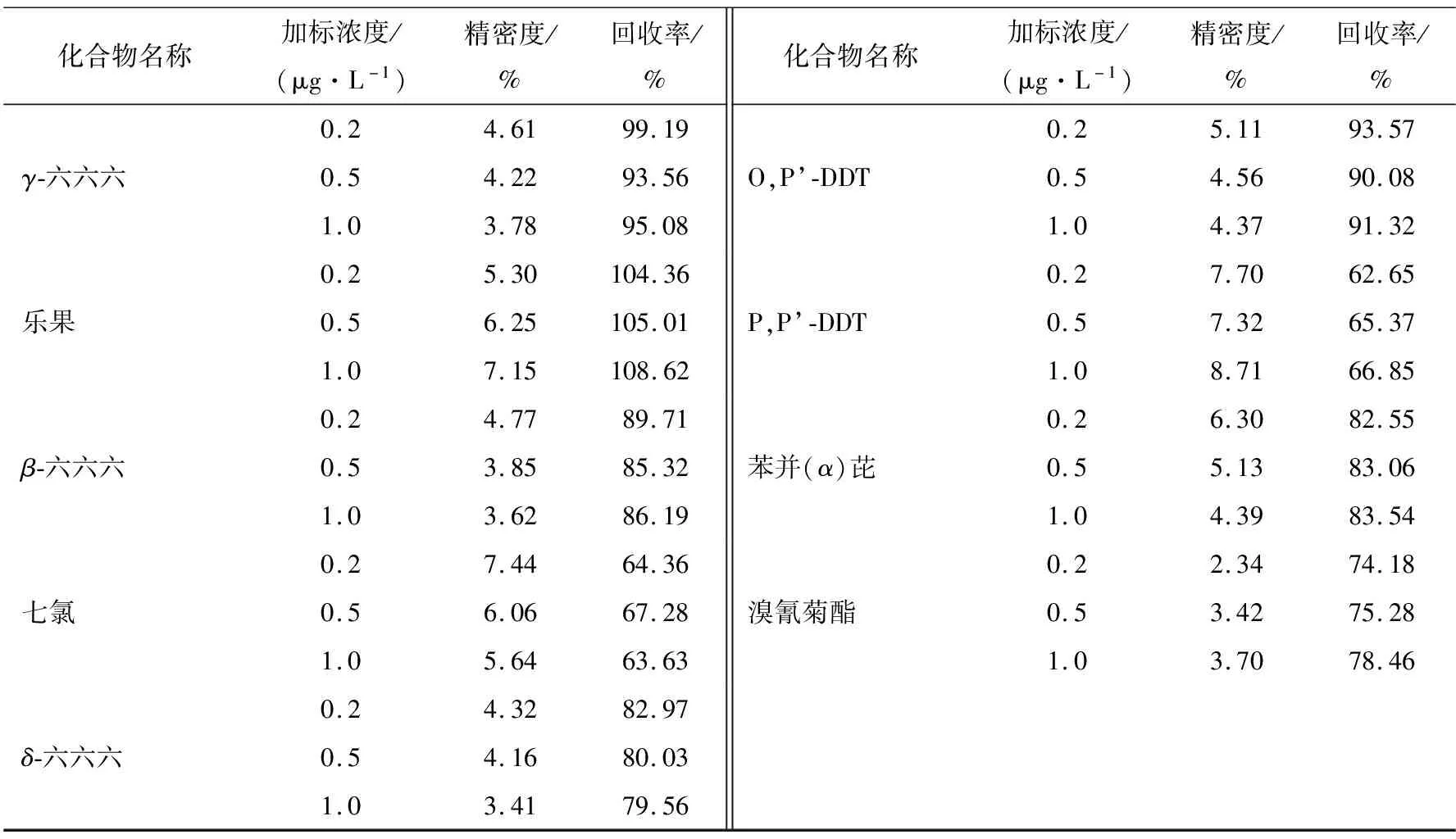
续表3
3 结论
建立了气相色谱—三重四级杆串联质谱仪检测饮用水中的SVOCs,实现了29种SVOCs的一次进样分析。采用小体积液液萃取方式将取样量减少至30 mL,简化了前处理步骤;通过大体积进样方式(进样量10 μL)降低了方法检出限;串联质谱的多反应监测模式,有效地提高仪器分析方法的信噪比。在0.2~10.0 μg/L 质量浓度范围内,各化合物的响应值与浓度呈良好的线性关系,方法检出限为0.003~0.110 μg/L,加标回收率为61.7%~108.6%,RSD为1.5%~8.7%,适用于饮用水中29种半挥发性有机物的同时测定。
前处理过程不能实现采样、萃取及富集于一体[21],实现快速在线检测。苯并(α)芘的检出限与GB 5749—2006《生活饮用水卫生标准》中限量指标相同,定量计算误差较大,可考虑增大样品取样量至100 mL,实现苯并(α)芘的准确定量计算。
