基于第一性原理计算H2S、CO2、CH4在Fe-MoS2上的吸附性能
2020-05-13刘建仪陈奕兆
刘建仪, 陈奕兆, 谢 泱, 袁 华, 刘 淼
(1. 西南石油大学 油气藏地质及开发工程国家重点实验室, 成都 610500; 2. 西南石油大学 石油与天然气工程学院, 成都 610500)
1引 言
世界上的高含硫气藏的储量不可忽略,尤其是在常规能源开发技术日渐完善下,安全高效的开发此类气藏具有很高的经济价值,同时回收的硫元素也可用于化学工业. 由于施工成本等各种因素,难以清洁井场中的含硫天然气,需要通过管道将其运输到远程净化设备中清洁. 在这个过程中,井场和输送管道将不可避免地溢出H2S和CO2,这极大地危害了周围环境的安全并腐蚀管道[1-3]. 因此,必须找到一种有效的方法来去除溢出的H2S,CO2. 目前,文献研究了Mo(110),TiC的吸附性能,以及α-Fe2O3(001)和TiO2对H2S的吸附和离解性能[4-7]. Leonardo Hadlich de Oliveira报道了NAY沸石上的H2S吸附[8]. Noushin Osouleddini发现改性石墨烯表面含有TCNE分子作为CO2和CH4的良好吸附剂[9]. 近年来,由于石墨烯等二维层状纳米材料的兴起,二硫化钼(MoS2)由于其优异的机械、电子和光学性质而受到许多研究人员的广泛关注[10-12],并且MoS2单层具有对许多气体敏感的特征,诸多学者在这方面进行了大量研究[11,13,14]. 赵世军和薛建明发现NO,NO2,SO2与MoS2的结合被认为是所有被考虑的气体分子中吸附能力最强[13]. 大量研究证实,与MoS2单层相比,掺杂的MoS2单层结构更能改善对气体分子的吸附性能[15,16],其中部分研究表明掺杂Fe,Pt,Si,Ni等原子显著改善MOS2的吸附性能[11,17-22]. 通过文献的调查,MoS2中Fe的吸附式掺杂结构研究较少,同时相关研究中对高硫气藏特有的气体(CO2和H2S)吸附计算尚未见有报道. 本文采取第一性原理计算了H2S,CO2,CH4气体在Fe-MoS2上的吸附能,电荷转移,电子密度差等相关参数,得到了较好的结果.
2计算参数的设置
所有的计算都是基于MS软件的DOML3模块[23]. 建立了4×4×1的超晶胞MoS2,为防止周期性计算引起超晶胞层之间的相互作用,在C方向加入10 Å真空层.在执行结构优化时,使用Perdew-Burke-Ernzerhof(PBE)的广义梯度近似(GGA)来处理电子之间的相互作用能[24],并且使用单个有效势替代内核电子(DFT semi-core Pseudopots),在内核处理中引入相对论效应. 采用双数值轨道基组和轨道极化函数(DNP)的基组设置,DNP数值设为4.4. 能量收敛精度设置为2×10-5/Å,内部原子间相互作用力不超过4×10-3Ha/Å,最大位移设为5×10-3Å,自洽场(SCF)收敛精度为1×10-5Ha. 在所有计算中都考虑了自旋极化效应. 布里渊区采用Monkhorst-Pack网格,K点为5×5×1[25].
Morphology模块计算表明MoS2最易暴露的面为(003),因此选择(003)面为研究对象. 在文章中计算参数定义如下:
气体分子在Fe-MoS2上的吸附能为:
Eads=EFe-MoS2/gas-EFe-MoS2-Egas
(1)
电荷转移表示为:
Qt=Qa-Qb
(2)
3结果与讨论
3.1H2S、CH4、CO2、MoS2、Fe-MoS2的结构
图1的(a)-(c)表明了H2S、CH4、CO2的分子结构. 基于Mulliken 方法,H2S分子中H原子带有0.073e的正电荷,S原子带有-0.174e的负电荷;CH4分子中H原子带有0.055e的正电荷,C原子带有-0.218e的负电荷;CO2分子中O原子带有-0.251e的负电荷,C原子带有0.502e的正电荷. (d)图表明了MoS2的结构.Mo-S键长为2.418 Å.
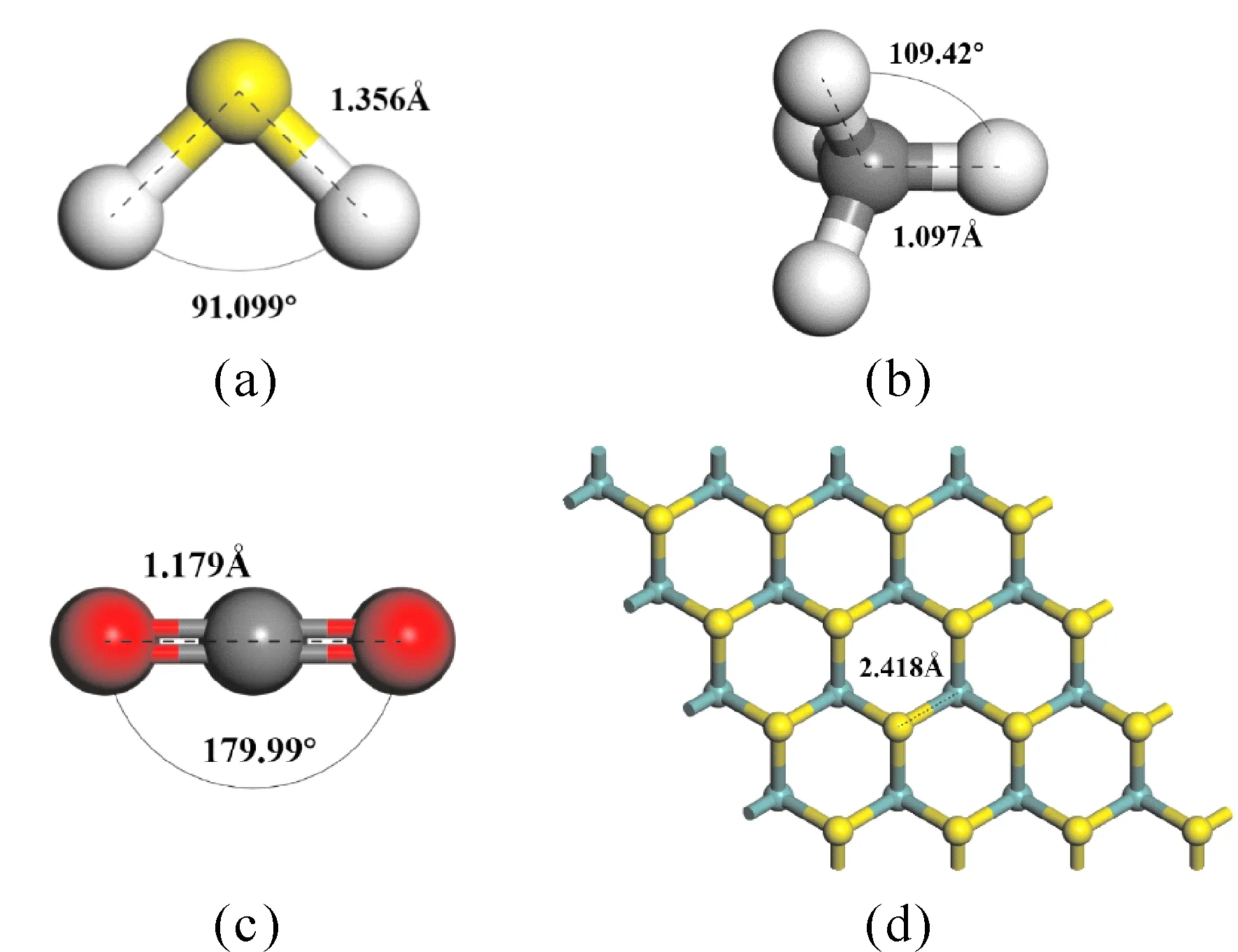
图1 H2S、CH4、CO2、MoS2的结构Fig. 1 The Structure of the H2S, CH4, CO2 and MoS2
考虑到MoS2材料的对称性,对于Fe吸附式掺杂在MoS2上存在三种可能的位置. Mo原子的顶部、在S原子的顶部、在MoS2六圆环的中心上方. 图2表示了这些位置,表1给出了吸附式掺杂后的相关吸附能. 计算结果表明Fe原子掺杂在MoS2中最稳定的位置是在Top-Mo,吸附能为-3.804eV.

图2 Fe掺杂在MoS2上的三种结构Fig. 2 Three structures of Fe doped on MoS2
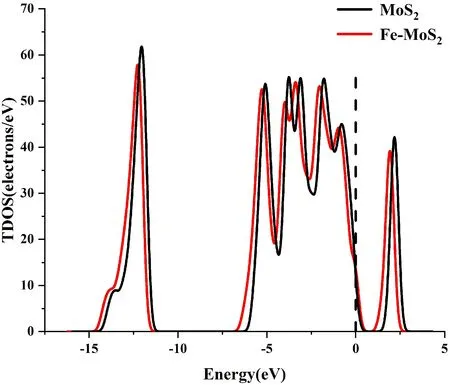
计算材料的总态密度(TDOS)和分波态密度(PDOS),定义费米能级EF=0 eV,进一步分析Fe原子掺杂在MoS2的掺杂机理. 图3所示,掺杂Fe原子后,材料的TDOS整体呈左移趋势,表明掺杂原子前后系统总能量发生了改变. 通过对PDOS波形分析,Mo原子的4d轨道与Fe原子的3d轨道在-5.5eV, -5eV, -3eV, -0.5 eV发生了重叠,表明Fe原子与Mo原子之间较强的轨道杂化作用.
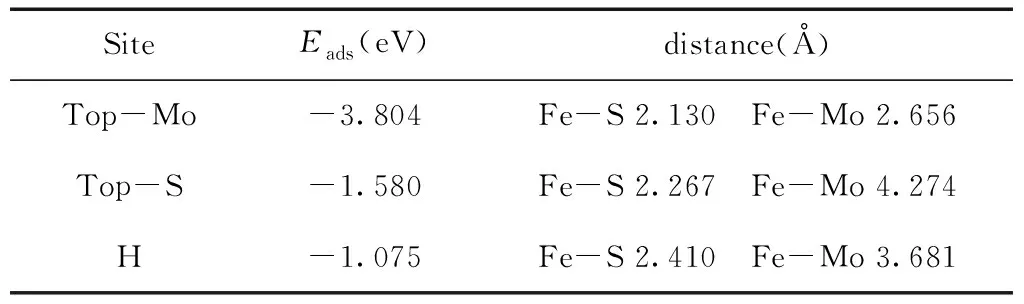
表1 不同掺杂位置的吸附能及距离
(a)
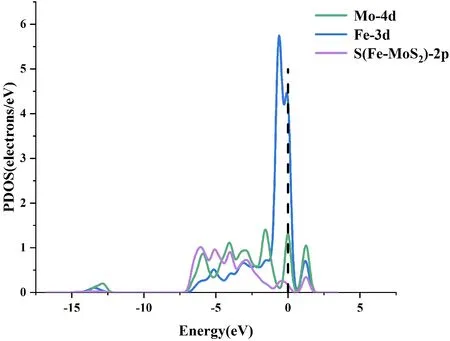
(b)图3 (a)掺杂前后体系TDOS; (b)Mo、Fe、S原子的PDOSFig. 3 (a) The TDOS of system doped before and after;(b)The PDOS of Mo, Fe and S
与此同时,也分别计算了Fe-MoS2和MoS2的带隙.带隙的减小,表明电子由价带激发到导带更加容易,体系更加稳定.

表2 掺杂前后材料的带隙
3.2H2S在Fe-MoS2上的吸附
进行了三次计算以验证H2S在Fe-MoS2上的吸附.M1,M3是H2S平行于Fe-MoS2,M2则是H2S垂直于Fe-MoS2. 这三种状态下的吸附参数都趋于一致. 表3给出了相关的参数信息,最高的吸附能量是M3系统,吸附能量为-1.512eV,吸附后H2S分子与Fe原子的距离为2.260Å. 分析气体分子吸附前后的总电荷数,气体分子的正电荷在吸附后增多,计算了Fe原子在吸附前后的电荷数,Fe原子电子增加. 表明在吸附过程中,电子由气体分子转移到Fe原子附近.

表3 H2S在Fe-MoS2上吸附的相关参数
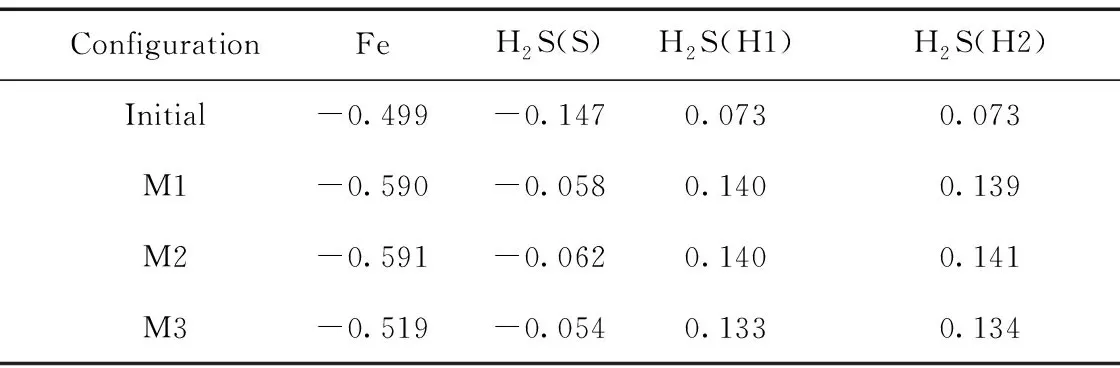
表4 吸附前后Fe原子和H2S分子电荷数
图4是优化后H2S分子吸附在Fe-MoS2的M3结构.吸附后,MoS2单层与H2S分子均发生了细微变化. 对于MoS2单层结构而言,Fe-Mo键长距离增大了0.066 Å,Fe-S(MoS2)的键长增大了0.023 Å,键长的增大表明了H2S的吸附使得Fe与MoS2的键合能力变弱. 对于H2S分子,键长略微增大0.002 Å,键角也较吸附前略微增加0.18°,表明吸附对气体分子结构影响很小.

图4 优化后H2S吸附在Fe-MoS2上的侧视图与俯视图Fig. 4 The side view and top view of optimized H2S adsorption on Fe-MoS2
图5是对H2S吸附在Fe-MoS2上的态密度图. 从TDOS图上显示,波形有轻微的左移,且在波峰处有所变化,表明吸附后材料发生了能量的变化. 对于PDOS, S原子的2p轨道与Fe原子的3d轨道在-4eV, -6eV, -7.5eV等位置有明显的波峰重叠,表明S原子与Fe原子发生了轨道杂化作用.

(a)
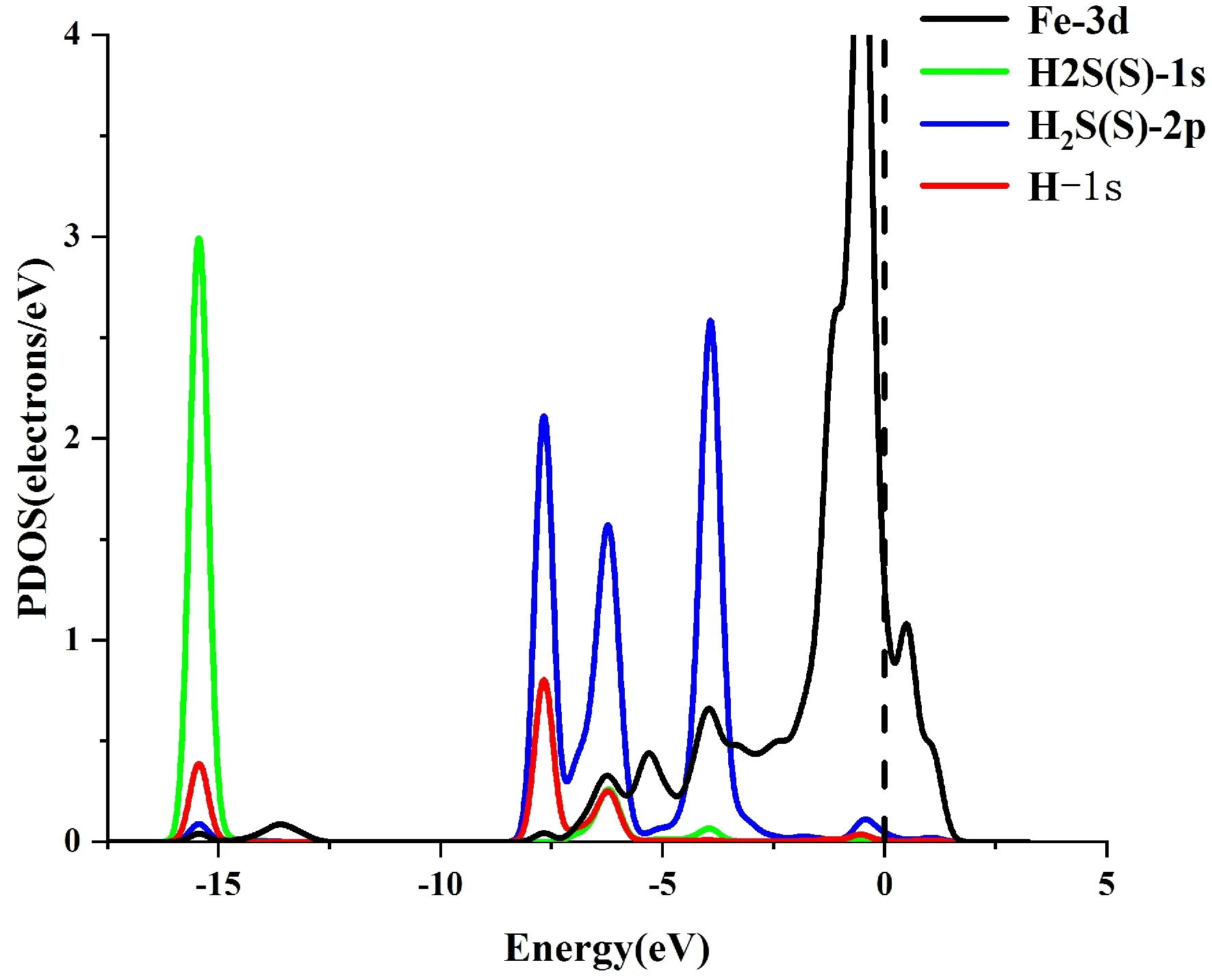
(b)图5 (a)吸附H2S前后的TDOS; (b)吸附H2S后的PDOSFig. 5 (a)The TDOS of system before and after adsorpting of H2S; (b)The PDOS of system after adsorpting of H2S
为了进一步分析H2S与Fe-MoS2之间的电荷转移机制,图6给出了Fe-MoS2吸附H2S的差分电荷密度图,H2S失去电子给Fe-MoS2,其中Fe原子的存在促进了H2S失去电子的能力.
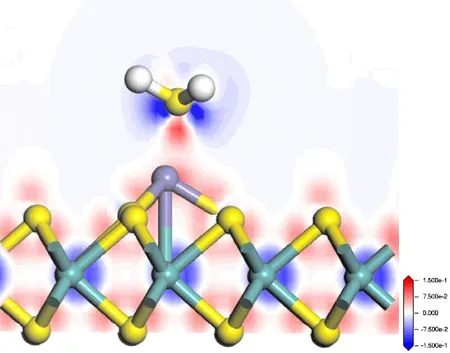
图6 H2S吸附在Fe-MoS2上的差分电荷密度Fig. 6 The differential charge density of H2S adsorbed on Fe-MoS2
3.3CH4在Fe-MoS2上的吸附
进行了三次计算以验证CH4在Fe掺杂在MoS2上的吸附,M1,M3是H原子垂直于Fe原子,M2是C原子垂直于Fe原子. 表5给出了相关的参数信息.最高的吸附能量是M1系统,吸附能量为-0.688eV.吸附能和电子转移都远远小于H2S在Fe-MoS2上的吸附参数. 表明这种吸附是一种较弱的作用力,属于物理吸附范畴.
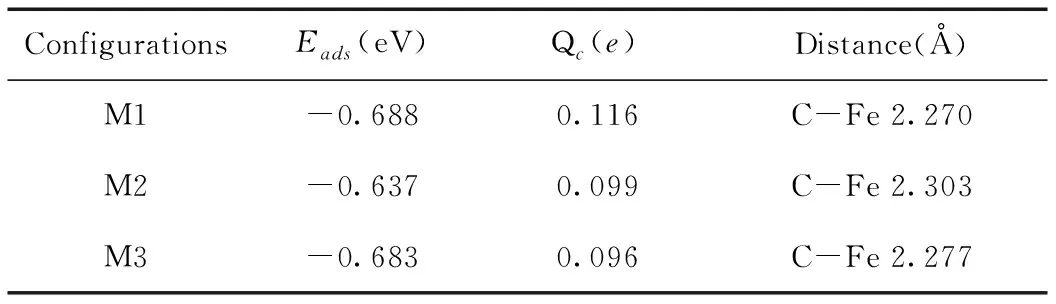
表5 CH4吸附与Fe-MoS2上的相关吸附参数
图7是优化完成的CH4吸附在Fe-MoS2的结构图. 吸附后CH4中C原子与Fe原子距离为2.270 Å. CH4分子的结构没有明显的变化,靠近Fe原子一侧的C-H键长略微增加了0.024 Å,另一侧的C-H键长保持不变. Fe原子与Mo原子之间的键长增加了0.075 Å,Fe原子与相邻S原子的键长增加了0.028 Å.这些略微的变化都表明这种吸附作用力十分微弱.

图7 优化后CH4吸附在Fe-MoS2上的侧视图与俯视图Fig. 7 The side view and top view of optimized CH4 adsorption on Fe-MoS2
图8给出了吸附后的TDOS与PDOS. 从TDOS中,吸附CH4前后体系的TDOS基本重合,表明吸附对体系没有产生能量的变化.从PDOS中,Fe原子与C原子和H原子重叠的部分很小,表明这种吸附不存在强力的相互作用,这与前面的数据分析结果相吻合.
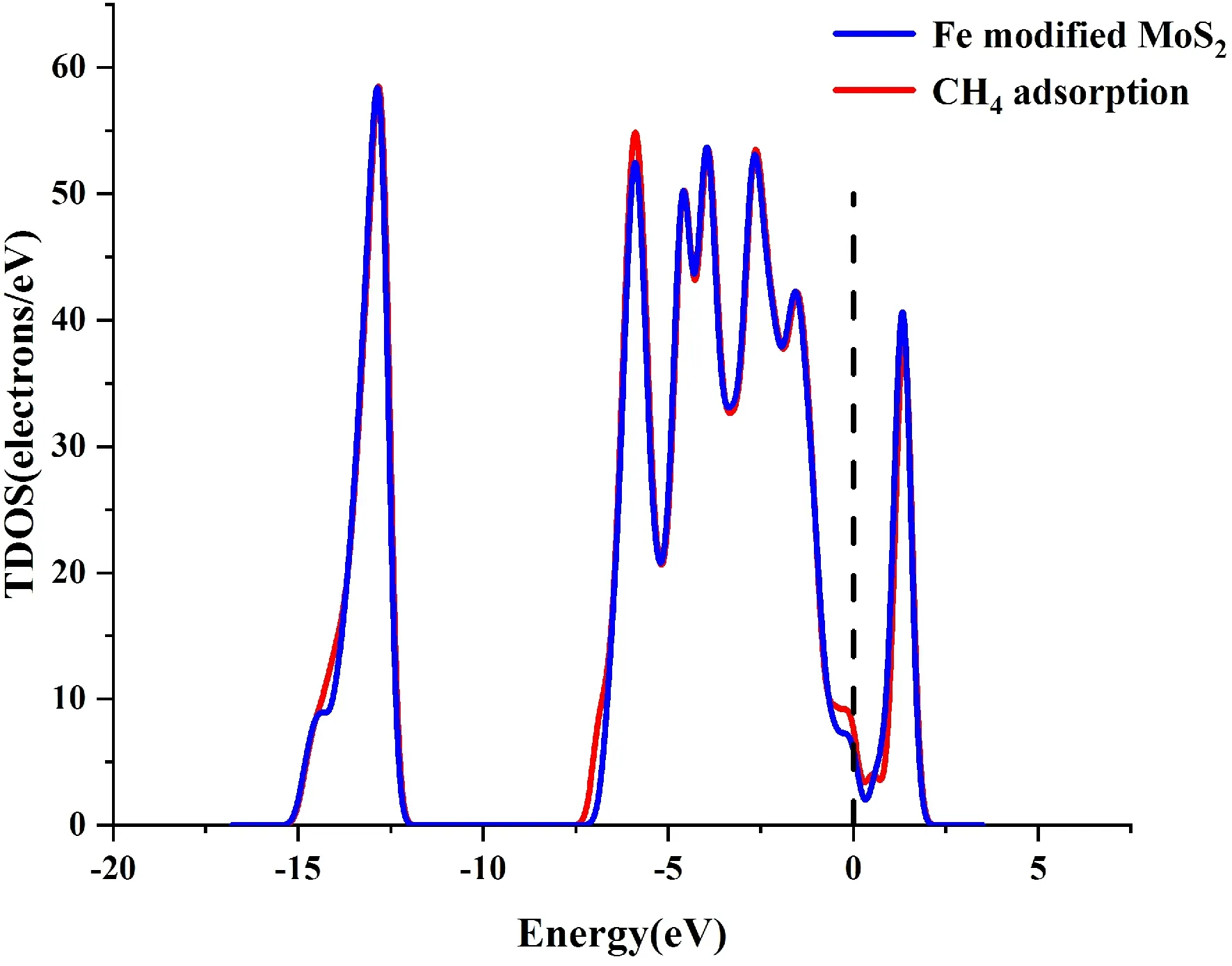
(a)
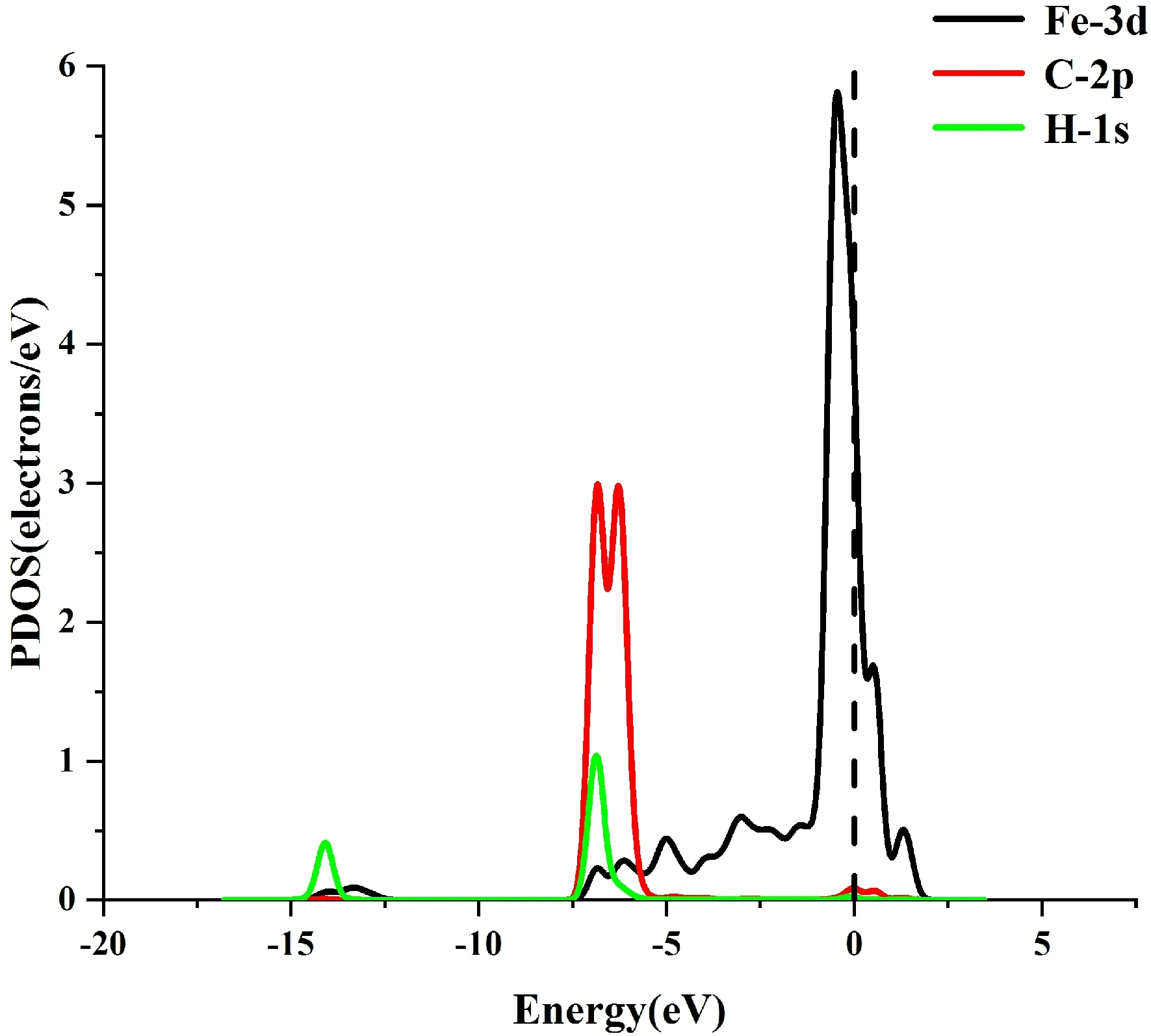
(b)图8 (a)吸附CH4前后的TDOS;(b)吸附CH4后的PDOSFig. 8 (a)The TDOS of system before and after adsorpting of CH4; (b)The PDOS of system after adsorpting of CH4
为了进一步分析CH4与Fe-MoS2之间的电荷转移机制,图9给出了Fe-MoS2吸附CH4的差分电荷密度,可以发现电子转移较弱. 表明CH4与Fe-MoS2之间相互作用力很弱,属于物理作用力.
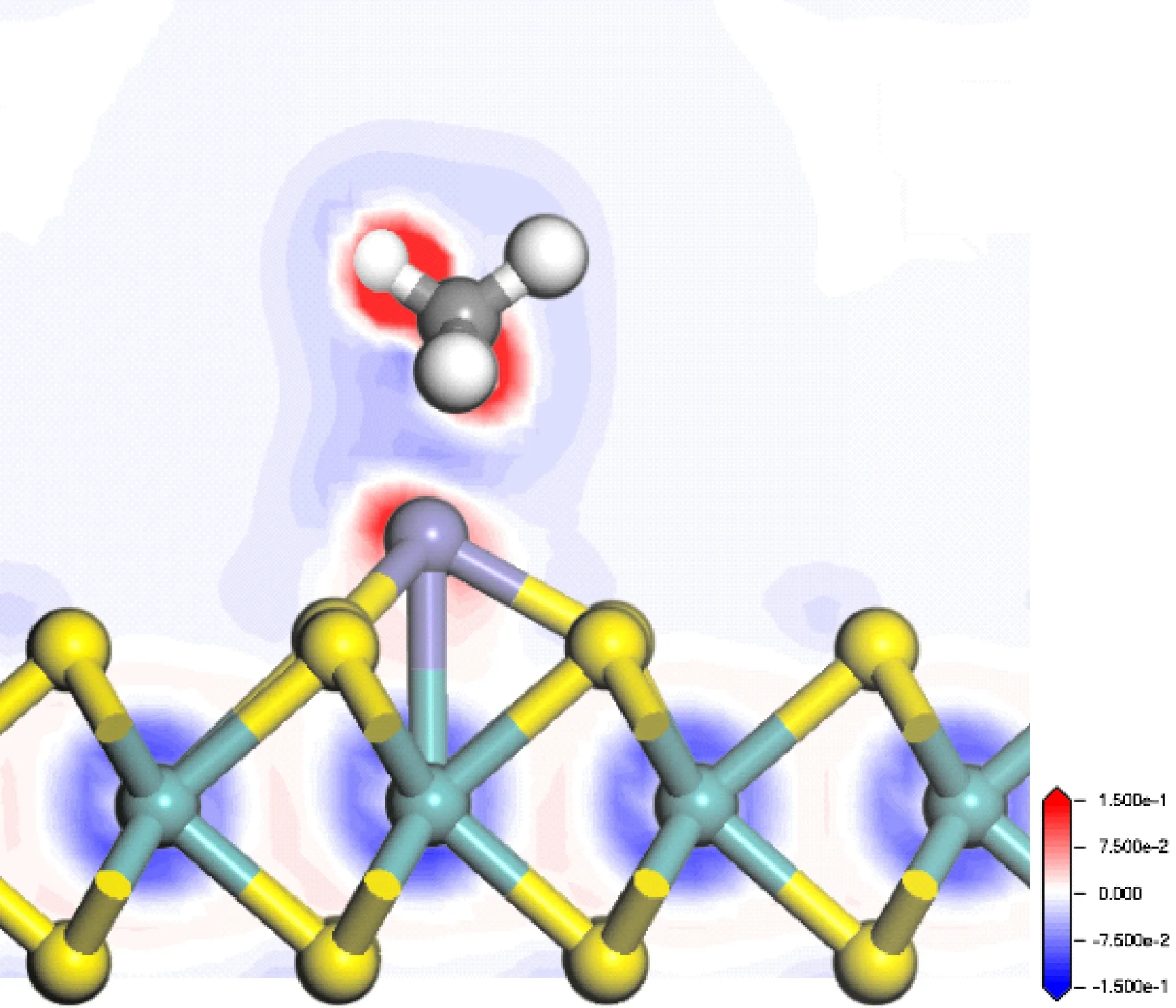
图9 CH4吸附在Fe-MoS2上的差分电荷密度图Fig. 9 The differential charge density of CH4 adsorbed on Fe-MoS2
考虑到CO2分子的对称性,在对CO2进行吸附计算时,在M1,M2系统中,CO2分子的O原子垂直于Fe原子上方,Fe原子的取向为O原子.M3,M4系统则是水平放置于Fe原子上方.
相关吸附参数已在表6中给出,最高的吸附能为-2.416 eV,较M1,M2的结果相对比,M3,M4的结果表明CO2在Fe-MoS2的吸附可能存在一种更加稳定,作用力更强的吸附. 从电荷转移也可以证实这点.
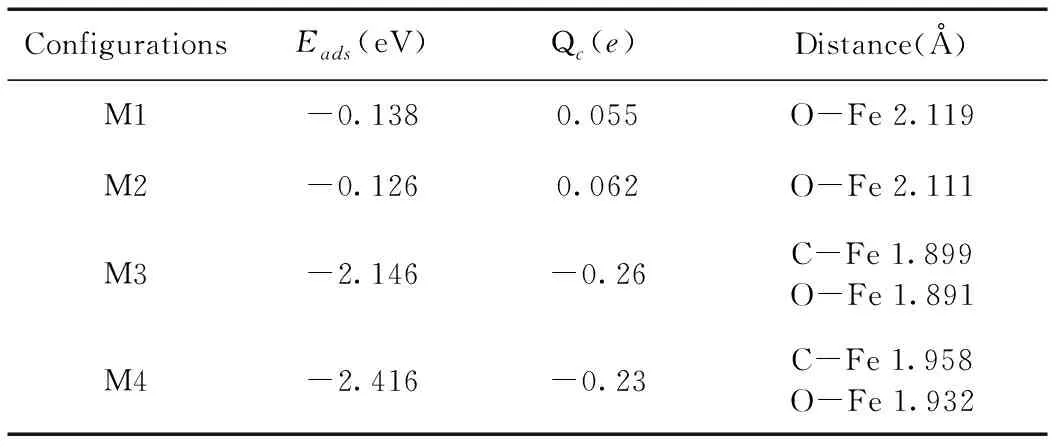
表6 CO2吸附在Fe-MoS2上的相关吸附参数
图10给出了M4吸附结构,CO2由直线型分子变化为V型分子,键角由179.99°减小到 143.654°,这表明着在C原子附近产生了大量的电子,电子的排斥作用使得键角发生了改变. Fe原子与O原子和C原子的距离分别为1.932 Å和1.958 Å. Fe原子与Mo原子的距离增加了0.156 Å,Fe原子与S原子的距离增加了0.057 Å,这些键长的变化表明了吸附作用比CH4与H2S分子的影响更加强烈. 从电子转移的角度来看,在M3与M4系统中,CO2是电子的受体,Fe原子是电子的给体,这与M1,M2系统恰好相反.

图10 CO2吸附在Fe-MoS2上的吸附结构侧视图与俯视图Fig. 10 The side view and top view of optimized CO2 adsorption on Fe-MoS2
图11给出了这种吸附结构的TDOS与PDOS.对于TDOS,在-8eV,-20.5 eV处发生了突变,这主要是O原子与C原子的2p轨道的贡献,表明了体系能量发生了变化.

(a)
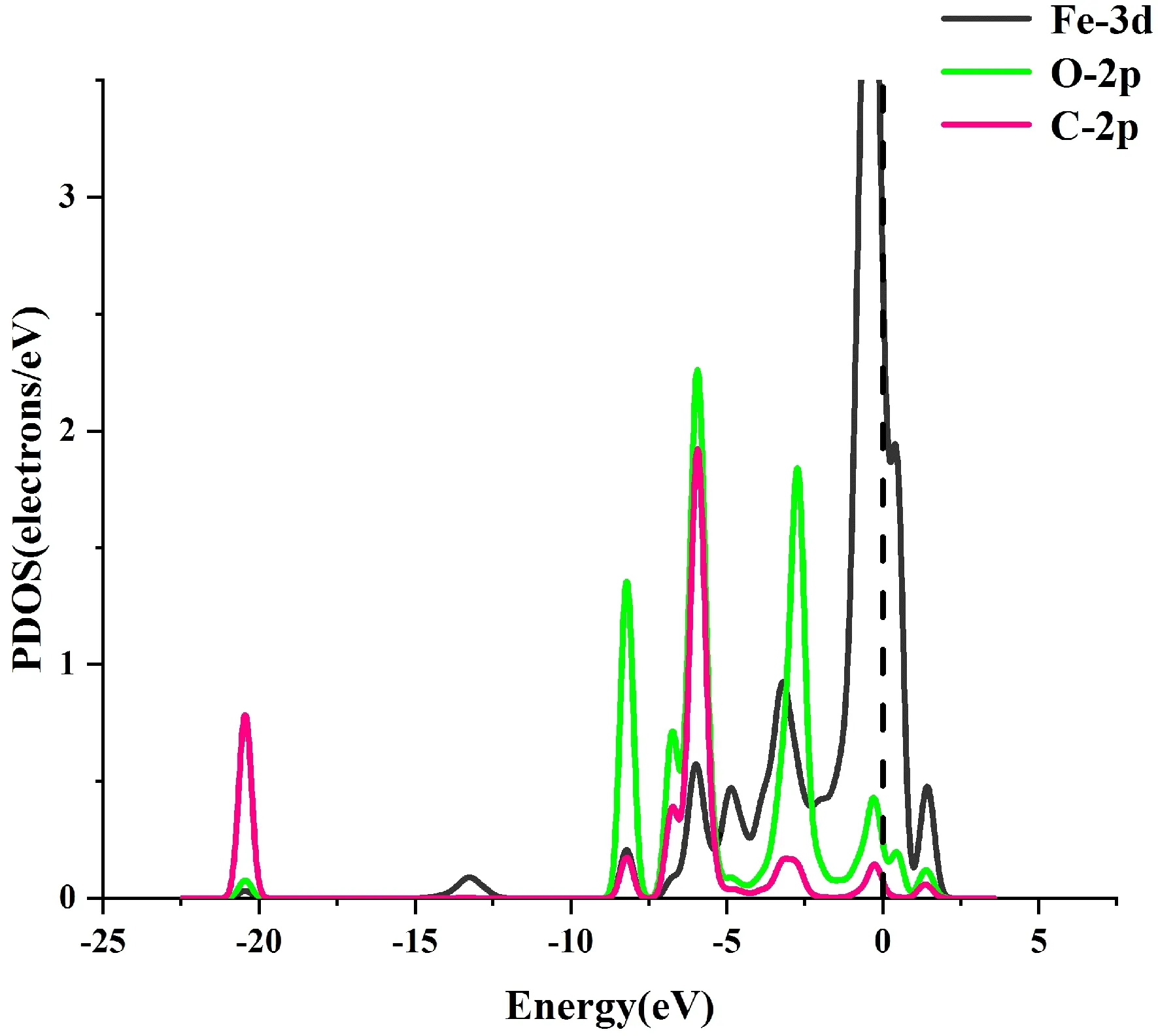
(b)图11 (a)吸附CO2前后的TDOS;(b)吸附CO2后的PDOSFig. 11 (a)The TDOS of system before and after adsorpting of CO2;(b)The PDOS of system after adsorpting of CO2
对于PDOS,在-4.5 eV,-5.5eV,-3eV,-2.5eV,-0.5 eV等能量处有峰的重叠,表明了Fe原子与C原子O原子之间存在轨道杂化作用.
图12给出了Fe-MoS2吸附CO2后的差分电荷密度图,CO2得到电子,Fe原子附近电子密度减小.这与前面的电子转移分析结果相吻合.
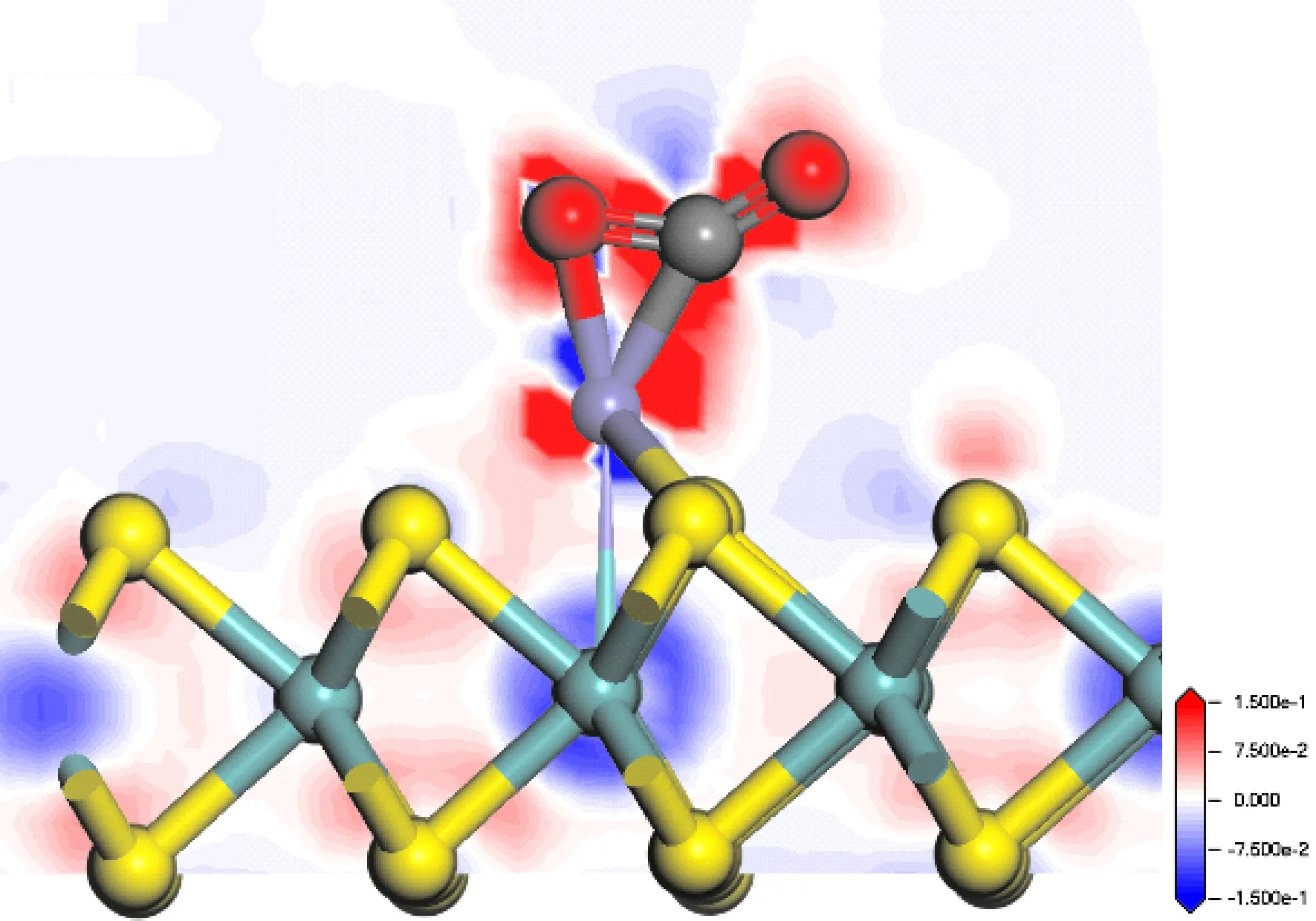
图12 CO2吸附在Fe-MoS2上的差分电荷密度图Fig. 12 The differential charge density of CO2 adsorbed on Fe-MoS2
4结 论
(1)计算了Fe原子在单层MoS2上存在的三种可能吸附的位点,得到了最稳定的吸附位置,位于Mo原子的顶部.
(2)分别计算了H2S,CH4,CO2三种气体在Fe-MoS2上的吸附结构,得到了相应的吸附参数.H2S,CH4,CO2在Fe-MoS2上的吸附能分别为-1.512eV,-0.688 eV,-2.416 eV.
(3)H2S和CO2在Fe-MoS2上是一种化学吸附,CH4在Fe-MoS2上是一种物理吸附.对于CO2在Fe-MoS2上的吸附,计算得到了一种较强的吸附结构.
(4)Fe原子掺杂MoS2的材料对三种气体有着不同的吸附特性,表明这种材料有可能被开发成为一种气体报警装置或是一种良好的气体吸附剂.
