端羟基聚丁二烯化学改性研究进展*
2020-05-13张平安张习龙袁剑民邓剑如
张平安,张习龙,袁剑民,邓剑如
(1.湖南大学 化学与化工学院,长沙 410082;2. 湖北三江航天江河化工科技有限公司,远安 444200)
0 引言
端羟基聚丁二烯(HTPB)是一种液体粘合剂,具有粘度低、低温及加工性能好、成本低廉等优点,是目前应用最广的固体推进剂粘合剂之一[1-3]。但由于HTPB主链结构中仅含有少量极性的羟基,使得该粘合剂存在极性小,与填料界面粘结弱等问题,从而限制了固体推进剂性能提高[4-5]。为提升固体推进剂的使用性能,推进剂配方中往往需要使用多种助剂,如键合剂、交联剂、燃烧催化剂等,但这些助剂往往会因为与粘合剂体系的热力学相容性不好,而制约其使用效果[6-9]。同时,HTPB作为惰性粘合剂,其能量水平较低,与极性增塑剂相容困难,在固体推进剂追求能量最大化的研究背景下,HTPB 也表现出一些不足[10]。随着推进剂的发展,对推进剂粘合剂也提出了更高的要求,各国均致力于新型粘合剂的开发和应用,但新型粘合剂往往由于成本高或难以规模化生产等原因导致应用研究仍处于探索阶段[11-13]。因此,HTPB在一定时间内仍为国内外固体推进剂用的主要粘合剂。为满足越来越高的工程应用要求,对传统HTPB进行化学改性,以提高其综合性能。
从分子结构上看,HTPB具有大量的不饱和双键及活性端羟基,这些结构为科研人员从源头改善HTPB的性能提供了可能。目前,对HTPB的改性主要通过以下四个途径实现:(1)端羟基的改性,将端羟基转化为其他端官能团;(2)末端碳原子上的功能化修饰;(3)对分子结构中双键的改性,引入功能性基团;(4)利用HTPB分子链段构筑嵌段共聚物。本文着重论述四种HTPB的改性方法及其研究进展。
1 HTPB的端羟基改性
HTPB分子链端带有活性较高的羟基,对其进行化学改性是提高其应用性能的有效方法。罗延龄等[14]采用官能团部分转化法,在一定条件下使HTPB与有机二酸酐通过酯缩合反应制备出带有端羟基和端羧基的聚丁二烯液体橡胶,使其同时兼备HTPB和CTPB(端羧基聚丁二烯)的双重功能,以改善HTPB推进剂压力指数高,对环境敏感性大的缺陷。同样,Barcia等[15]利用HTPB与马来酸酐反应制备了端羧基聚丁二烯CTPB,并在此基础上与双酚A缩水甘油醚(DGEBA)反应制备出端环氧基聚合物,合成路线如图1所示。改性后的HTPB提高了与环氧树脂基体之间的相容性,并提供更好的界面粘接性。柏海见等[16]通过化学改性的方法将HTPB上的羟基转化成胺基,从而合成了端胺基聚丁二烯,合成路线如图2所示。由于端胺基比羟基的活性更高,因此与异氰酸酯固化剂反应速率更快,可提高HTPB的固化性能。
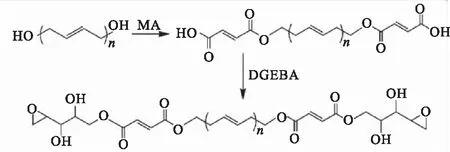
图1 端环氧聚丁二烯环氧树脂的制备Fig.1 Preparation of the polybutadiene-epoxy resin

图2 端胺基聚丁二烯的制备Fig.2 Preparation of the amino terminated polybutadiene
为克服传统聚氨酯粘合剂在制备过程中对环境湿度敏感的缺点,黄进等[17]通过 HTPB与3-溴丙炔在碱性条件下发生缩合反应,制备了端炔基聚丁二烯(PTPB),并将 PTPB 与叠氮缩水甘油醚(GAP)混合,利用点击化学反应在室温条件下实现固化交联,反应路线如图3所示。制备的弹性体与聚氨酯弹性体相比,因其分子结构中引入了体积较大的三唑环,影响了主链的柔顺性,其伸长率(37.3%~87.7%)低于聚氨酯弹性体(150%~900%),但模量得到显著提升(GAP/PTPB弹性体模量为0.65~5.36 MPa,聚氨酯为0.05~1.9 MPa)。同时,化学交联提高了PTPB和GAP的相容性,其固化过程不受环境湿度等因素的影响。杨荣杰等[18-19]同样探讨了利用点击化学反应制备不受环境水分影响的聚丁二烯弹性体。制备过程首先将HTPB转化成端氯基聚丁二烯(ClTPB),然后再与NaN3反应制备端叠氮基HTPB(ATPB,图4)。进一步以三炔丙基胺为固化剂,通过点击化学反应实现叠氮基与炔基之间的固化反应。测试结果表明,当R值为1.0时,弹性体拉伸强度为0.42 MPa,断裂伸长率为365%。
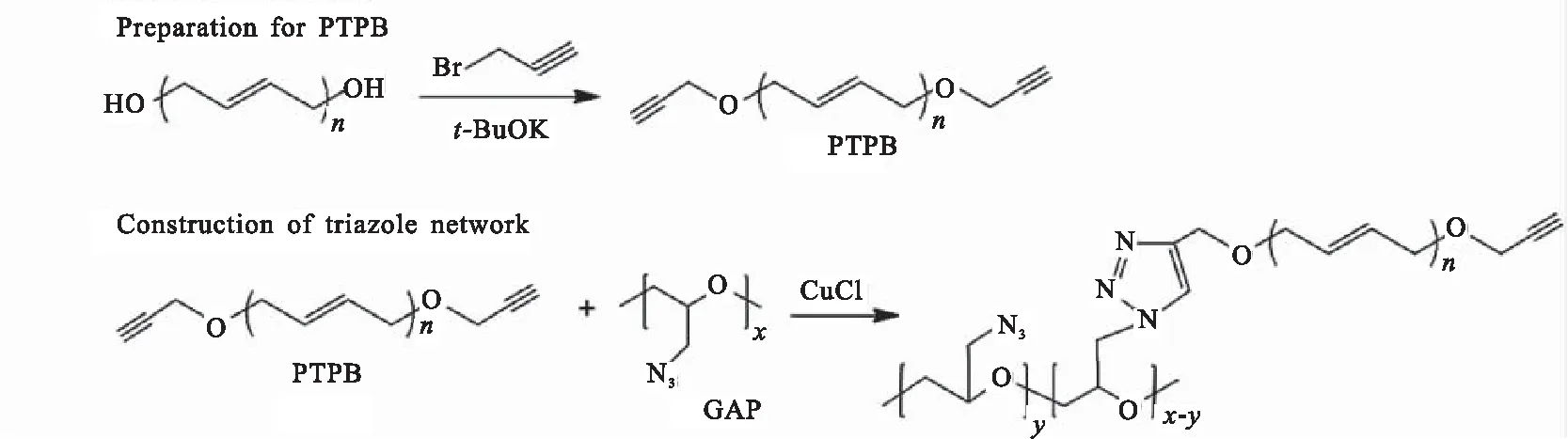
图3 PTPB的制备及固化交联示意图Fig.3 Preparation of PTPB and construction of crosslinked network

图4 ATPB的制备及固化交联示意图Fig.4 Preparation of ATPB and construction of crosslinked network
针对端炔基聚丁二烯与叠氮聚合物及小分子交联剂相容性差,易发生相分离的问题,Reshmi等[20]通过对HTPB端羟基进行化学改性,分别合成了含氨基甲酸酯结构单元的端炔基聚丁二烯(PrTPB)及端叠氮基聚丁二烯(AzTPB,图5),最后通过点击化学反应实现了二者的固化交联。结果表明,固化后的弹性体力学性能要明显优于HTPB-TDI聚氨酯弹性体。当R值为1.0时,叠氮-炔基弹性体拉伸强度为1.52 MPa,断裂伸长率为660%(HTPB-TDI弹性体拉伸强度为0.86 MPa,断裂伸长率为240%)。装药实验结果表明,使用该新型粘合剂的推进剂力学性能较HTPB粘合剂提升了14%~22%。

图5 PrTPB及AzTPB的制备Fig.5 Preparation of PrTPB and AzTPB
Li等[21]同样采用两步法制备了含氨基甲酸酯结构的端炔基聚丁二烯(PUPB,图6),并与自制的三官能度叠氮交联剂反应制备了力学性能优良的弹性体。与Reshmi等[20]的合成路线相比,该方法避免了HTPB与TDI产生的扩链现象,产物的分子结构可控。研究表明,分子结构中氨基甲酸酯结构单元之间通过氢键相互作用,可进一步增强弹性体的力学性能。相比于不含氨基甲酸酯结构的PTPB(图3),其拉伸强度由0.62 MPa提升至4.2 MPa,断裂伸长率由47%提升至81.5%。且经30%质量的癸二酸二辛酯增塑后,断裂伸长率可提高至293.3%,拉伸强度降至1.31 MPa。

图6 PUPB的制备Fig.6 Preparation of PUPB
基于HTPB的叠氮-炔基固化体系具有环境湿度不敏感、反应条件温度、反应高效可控的特点。极性基团的引入可提高与含能增塑剂的相容性,增强与氧化剂填料的界面结合力,更重要的是可提高推进剂能量。
2 HTPB的末端碳原子改性
HTPB主链末端的碳原子也可通过化学反应进行功能化修饰,Sankar等[22]将1-氯-2,4-二硝基苯(DNCB)通过亲核取代反应接枝至HTPB末端碳原子上,得到含有硝基的HTPB-DNCB(图7)。通过IR、NMR、UV-vis等检测方法,证明了该反应发生在HTPB末端碳原子上,并对反应机理进行了解释:反应过程中NaH可夺取HTPB的末端羟基上的质子,并产生氧阴离子。氧阴离子不稳定,因此从相邻的碳原子上夺取质子,使该碳原子转化为烯丙基阴离子,该阴离子在反应过程中充当亲核试剂;而DNCB分子中含有强吸电子的硝基,使得氯原子较容易离去,故 DNCB分子可通过亲核取代反应连接在HTPB末端碳原子上(图8)。
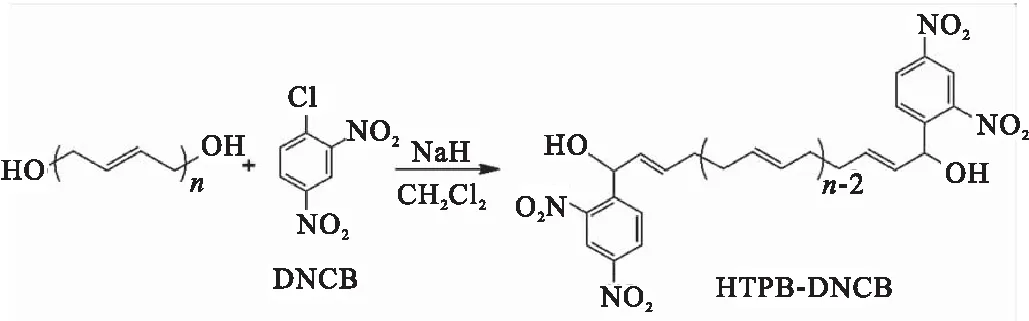
图7 HTPB-DNCB的制备Fig.7 Preparation of HTPB-DNCB

图8 HTPB与DNCB反应机理Fig.8 Reaction mechanism for HTPB and DNCB
为进一步增加HTPB的能量水平,Sankar等[23]利用三聚氯氰与HTPB末端碳原子反应制备中间体HTPB-CYC,然后再与NaN3反应向分子中引入叠氮基(HTPB-DT,图9)。叠氮基的引入不仅提升了粘合剂的整体能量水平,且在固化过程中可与氨基甲酸酯单元相互作用而形成微相分离,显著提高HTPB-DT基聚氨酯弹性体的力学性能[24]。

图9 HTPB-DT的制备Fig.9 Preparation of HTPB-DT
另外,针对二茂铁燃速催化剂数均相对分子质量低,与HTPB相容性差而导致的迁移、挥发,影响贮存稳定性等问题,Rao等[25]将二茂铁催化剂接枝到HTPB末端碳原子上(Fe-HTPB-1,图10),并将改性后HTPB作为粘合剂用于复合固体推进剂的制备中,对药柱贮存稳定性和燃烧速率进行了研究。结果表明,末端碳原子改性对HTPB微观结构影响较小,产物粘度较HTPB变化不大。推进剂在高温和低温环境均表现出良好的稳定性,二茂铁分子没有发生聚集和挥发,且药柱燃烧速率提高 125%。

图10 Fe-HTPB-1的制备Fig.10 Preparation of Fe-HTPB-1
通过HTPB末端碳原子对HTPB进行改性,在实现功能化的同时保留了末端羟基,用于推进剂粘合剂时对固化体系影响较小。同时,末端碳原子改性并没有改变HTPB的微观结构,因此改性后依然保留原HTPB的优良物理化学性能。
3 HTPB的双键改性
通过对HTPB分子结构中的双键进行改性也是提高HTPB性能的另一个重要途径,其中国内外报道较多的是HTPB的环氧化改性。国外早在20世纪50年代便开展了HTPB环氧化改性研究,Greenspan等[26]以过氧乙酸为氧化剂与HTPB进行反应制备了环氧基质量分数为6.6%的环氧化端羟基聚丁二烯(EHTPB)。Choi等[27]用间氯过氧苯甲酸(m-CPBA)原位法合成了一系列不同环氧值的EHTPB。研究表明,环氧化程度(物质的量分数)每增加1%,EHTPB的玻璃化温度增加0.8 ℃。研究还发现主链上的双键反应活性不同,其大小顺序为[顺l,4-双键]> [反1,4-双键]> [1,2-双键]。Nikje等[28-29]利用过硫酸氢钾复合盐(杜邦商品名为 Oxone)与丙酮作用生成的二甲基双环氧乙烷为氧化剂,对HTPB进行环氧化改性,反应式如图11所示。作者对结构中三种双键的反应活性进行了研究,其结论与Choi[27]一致。
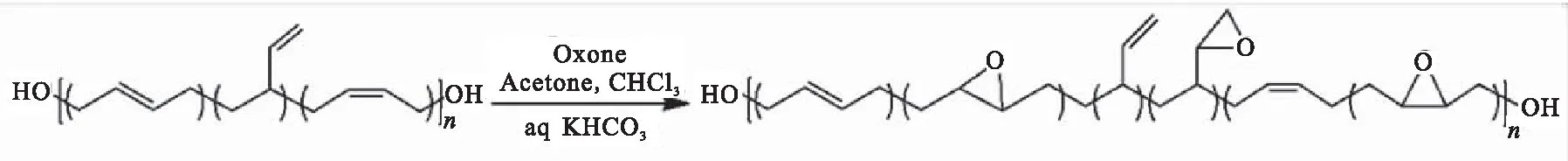
图11 EHTPB的制备Fig.11 Preparation of EHTPB
国内张小华等[30]采用过氧甲酸原位法对HTPB进行了环氧化研究,详细探讨了反应温度、时间和原料配比等因素对反应的影响,并对环氧化产物的固化性能进行了研究。研究表明,EHTPB可用胺类或酸酐类固化剂固化,与环氧树脂具有较好的相容性,可显著提升固化物的柔韧性和耐热性。郑元锁等[31-32]利用过氧乙酸原位氧化法对不同数均相对分子质量的HTPB进行环氧化改性,系统研究了反应温度、时间、物料比等因素对产物环氧值、开环率的影响,并对环氧化反应动力学进行了研究。在后续研究中分别以H12MDI和TDI为固化剂,深入探究了EHTPB的固化反应动力学及弹性体的力学性能[33-34]。结果表明,EHTPB型聚氨酯力学性能优于HTPB型聚氨酯材料,但其固化温度较高(70 ℃),限制了EHTPB在推进剂粘合剂中的应用。为此,刘磊等[35]对EHTPB的低温固化工艺进行了探究,最终使用钛酸酯偶联剂(TC-114)在50 ℃实现固化,其固化产物的拉伸强度达到0.75 MPa,断裂伸长率达到110%。
与HTPB相比,EHTPB分子中引入的环氧基团为高反应活性官能团,可作为新的反应中心进一步改性。为克服HTPB能量水平低的缺陷,Colclough等[36]首先采用过氧甲酸原位法制备EHTPB,将得到的EHTPB在有机溶剂中与 N2O5反应,得到邻位的二硝酸酯聚合物NHTPB-1(图12)。研究表明,硝化程度对产物理化性能影响较大,当硝化度为10%时(HTPB中10%的双键转化为硝酸酯基),此时产物的粘度为12.8 Pa·s,Tg为-58 ℃;当硝化度为20%时,粘度增至200 Pa·s,Tg升至为-22 ℃。硝化度为10%的产物可较好的平衡能量与推进剂力学性能之间的关系,且与含能增塑剂具有较好的相容性。作者还将硝化程度为10%的NHTPB-1与部分含能粘合剂的理化性质做了对比[37](表1),发现NHTPB-1的性能与PolyNIMMO、PolyGLYN相近,但成本更低,具有较好的应用前景。

图12 NHTPB-1的制备Fig.12 Preparation of NHTPB-1

表1 NHTPB-1与部分含能粘合剂理化性能对比
Note∶Mnof PolyNIMMO,PolyGLYN,and 10%NHTPB-1 are 2000~15 000,1000~3000,2500.
王庆法等[38]采用相同的合成路线制备了不同硝化度的NHTPB-1,考察了硝化度对Tg和热稳定性的影响。结果表明,随着硝化度的增加,Tg呈线性增加,热稳定性逐渐降低。当硝化度较低时(<15%),产物具有较好的热稳定性。王勃等[39]则采用稀硝酸对环氧化程度为10%的EHTPB进行开环,合成了硝酸酯基HTPB(NHTPB-2,图13)。结果表明,产物具有良好的安全性能,与硝酸酯含能增塑剂容性好,采用NHTPB-2粘合剂的推进剂配方比HTPB 粘合剂配方燃速提高了2 mm/s。

图13 NHTPB-2的制备Fig.13 Preparation of NHTPB-2
Pant等[40]则利用双键易于加成的特点,采取两种不同的路线合成了叠氮化HTPB(Azido-HTPB),其合成路线如图14所示。虽然路线2中乙酸锰(III)催化叠氮化反应作为一步合成法似乎更为方便,但却存在反应结果重复性差、产物难以分离纯化等问题,而路线1可通过控制溴的添加量精确控制HTPB的改性程度和产物中叠氮基的含量。作者对不同改性程度的产物粘度和Tg进行检测,发现产物粘度和Tg均随着改性程度的增加而升高。当HTPB改性程度为10%(双键百分比)时,产物粘度为11 Pa·s,Tg为-66℃,满足推进剂粘合剂使用要求。Ghayeni等[41]报道了一种通过一步反应,同时向HTPB双键结构中引入硝基和叠氮基的方法(Azide-nitrato-HTPB,图15),并探讨了反应温度、时间等对改性程度的影响,制备的产物中叠氮基含量为7.15%,硝基含量为5.17%,产物粘度为17.6 Pa·s。该反应效率较低,需在50 ℃下反应5~7 d,目前还难以对其进行放大和应用研究。

图15 Azide-nitrato-HTPB的制备Fig.15 Preparation of Azide-nitrato-HTPB
上述改性方法在引入含能基团时会破坏HTPB双键结构,进而影响其性能。Pant等[42]报道了一种以亚硝酸钠、碘为底物,通过一步反应制备硝基HTPB(Nitro-HTPB,图16)的方法。反应过程中,亚硝酸钠与碘反应生成活性物质NO2I,NO2I选择性的加成到HTPB双键结构中生成硝基碘化合物,最后可经碱处理脱除HI还原HTPB的双键。制备的产物中硝基含量为HTPB双键的10%~15%,并具有良好的热稳定性。Abusaidi等[43]采用上述方法制备了Nitro-HTPB,并作为粘合剂应用于推进剂配方中。结果表明,该粘合剂与含能增塑剂表现出良好的相容性,在添加含能增塑剂Bu-NENA(添加量3%)情况下,药柱仍具有较好的力学性能,且推进剂燃烧速率提高了40%;同时,对撞击和摩擦感度进行了测试。结果表明,Nitro-HTPB推进剂的撞击感度和摩擦感度较丁羟推进剂略有增加,但仍在安全范围内。
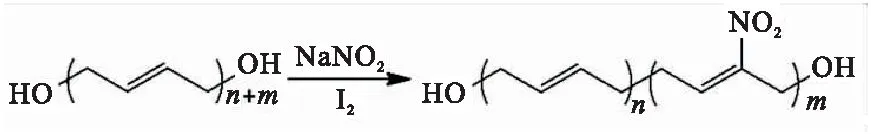
图16 Nitro-HTPB的制备Fig.16 Preparation of Nitro-HTPB
小分子增塑剂在粘合剂中易迁移、析出,有研究人员提出在推进剂配方中用数均相对分子质量较低的含能聚合物充当增塑剂,且聚合物增塑剂与粘合剂结构越相似,其相容性就越好[44-45]。为此,Ashrafi等[46]采用亚硝酸钠-碘硝化法对低数均相对分子质量的聚丁二烯(Mn=2670)进行改性,制备出硝基聚丁二烯(Nitro-PB),并将该产物作为含能增塑剂用于Nitro-HTPB粘合剂体系中。研究结果表明,Nitro-PB聚合物增塑剂数均相对分子质量(Mn=3000)远高于小分子增塑剂,使其在粘合剂中扩散速率较低。同时,NHTPB/NPB固化体系拉伸强度较HTPB/DOA体系有所增加,但断裂伸长率不及后者。这是由于体系中硝基含量的增加,导致分子间的相互作用力增强,作者提出可通过降低Nitro-PB的数均相对分子质量,进一步提高其力学性能。
通过化学反应将二茂铁燃速催化剂接枝到HTPB主链上,以解决二茂铁在贮存过程中易迁移的问题,也是目前HTPB双键改性的研究热点。法国火炸药协会(SNPE)将二茂铁基硅烷衍生物接枝到端羟基聚丁二烯上,得到了一种新型粘合剂型燃速催化剂,命名为Butacene[47],其结构式如图17所示。与小分子二茂铁相比,其最突出的优点是不会挥发,不向包覆绝热层界面迁移,且药浆工艺性能好。但其合成路线较为复杂,科研人员在此基础上提出了多种改进方法。

图17 Butacene结构式Fig.17 The structure of Butacene
Saravanakumar等[48]采用AlCl3为催化剂,通过Friedel-Crafts烷基化反应将二茂铁接枝到HTPB上,制得一种不迁移的二茂铁化合物Fc-HTPB-2,制备过程如图18所示。该路线仅需一步就实现了二茂铁接枝到HTPB上。研究发现,当二茂铁的质量分数达到2 %时,接枝聚合物粘度迅速上升,几乎不溶于任何溶剂,导致其在实际应用中产生潜在的不良影响。Subramanian等[49]以AIBN为引发剂,通过自由基加成反应将乙烯基二茂铁接枝到HTPB不饱和双键上(图19)。研究发现固化后的推进剂在100 ℃条件下人工加速老化50 h未发现迁移现象,但该方法的缺点是副反应较多且乙烯基二茂铁的接枝率非常低(仅为0.43%)。
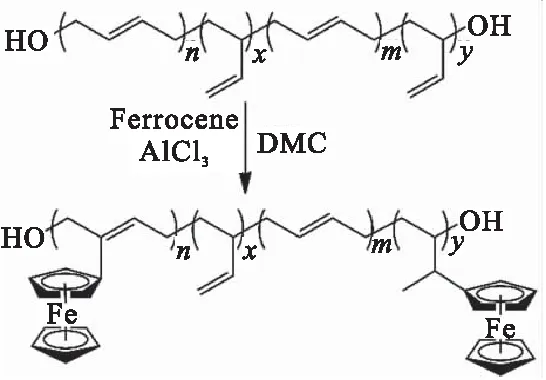
图18 Fe-HTPB-2的制备Fig.18 Preparation of Fe-HTPB-2
图18 Fe-HTPB-2的制备Fig.18 Preparation of Fe-HTPB-2

图19 乙烯基二茂铁接枝HTPB反应Fig.19 Synthesis of polyvinyl ferrocene grafted HTPB
国内张岩等[50]采用四丁基溴化铵作相转移催化剂,将燃速催化剂二茂铁甲酸接枝在EHTPB 的分子链上(图20),其平均接枝率达到了20.65%,解决了二茂铁燃速催化剂的迁移问题,但其铁含量受EHTPB环氧值的制约。为进一步提高二茂铁接枝HTPB中铁含量,张敏等[51]在制备出有机硅氢双核二茂铁衍生物基础上,通过铂催化剂将双核二茂铁接枝HTPB双键上,其结构式如图21所示。所得目标产物的铁含量为8.24%,产物的接枝率为63%。

图20 二茂铁甲酸与EHTPB的接枝反应Fig.20 Grafted reaction of ferrocene garboxylic acid with EHTPB

图21 双核二茂铁接枝HTPB目标产物分子结构Fig.21 The structure of binuclear ferrocene grafted HTPB
通过HTPB分子结构中的双键进行化学改性而实现其功能化,具有其独特的优势,但也存在一些难以避免的问题。如改性过程会破坏 HTPB 结构中的双键,而微观结构的改变将导致其理化性能的改变。因此,双键改性 HTPB往往存在低温性能下降,粘度增加等问题。
4 HTPB的嵌段共聚改性
HTPB 的嵌段共聚改性是利用 HTPB 的活性端羟基引发聚合反应,将具有特定功能的其他聚合物连接在 HTPB 的两端形成嵌段共聚物。Vasudevan等[52]以HTPB为大分子引发剂,BF3·OEt2为催化剂,引发环氧氯丙烷进行阳离子开环聚合,生成三嵌段共聚物PECH-PB-PECH,然后与叠氮钠反应制备叠氮缩水甘油醚-聚丁二烯-叠氮缩水甘油醚(GAP-PB-GAP)三嵌段共聚物(图22)。该共聚物中叠氮基团的存在使其可以作为高能粘合剂用于固体火箭推进剂,与AP/HTPB推进剂相比,该共聚物粘合剂体系可提高推进剂的燃烧速率,改善界面粘接性能。Reddy等[53]同样以HTPB为引发剂,在BF3·OEt2催化剂作用下引发BAMO单体开环聚合,生成三嵌段共聚物BAMO-HTPB-BAMO(图23)。测试结果表明,该嵌段共聚物具有良好的力学性能,且通过共聚的方法解决了含能粘合剂与HTPB难以相容的问题,进一步拓宽了HTPB粘合剂的使用范围。Cappello等[54]采用相同的思路,以HTPB为引发剂,对四种卤代、甲苯磺酰化单体进行开环聚合,制备了不同结构的HTPB-GAP嵌段共聚物(图24)。研究发现,反应过程中添加抗老化剂可抑制叠氮基与HTPB双键的自交联反应,但反应过程时间较长,其中聚合过程需要120 h,叠氮化过程需要在96 ℃下反应22 h,且产物需低温、避光存储。

图22 GAP-PB-GAP嵌段共聚物的制备Fig.22 Preparation of GAP-PB-GAP block copolymer

图23 BAMO-HTPB-BAMO嵌段共聚物的制备Fig.23 Preparation of BAMO-HTPB-BAMO block copolymer

图24 HTPB-GAP嵌段共聚物制备路线Fig.24 Preparation routes of HTPB-GAP block copolymer
Filippi等[55]则提出了一种反应更迅速的HTPB-GAP嵌段聚合物制备方法。制备过程中采用六亚甲基二异氰酸酯(HDI)或己二酰氯作为扩链剂,直接将HTPB与GAP通过扩链剂桥接在一起,形成HTPB-GAP嵌段聚合物,以HDI为例,其反应式如图25所示。与阳离子开环聚合的方法比较,该方法可快速获得HTPB-GAP的嵌段聚合物,缺点是该反应容易出现凝胶现象,反应对水敏感,需严格除水,最终的产物无法纯化。
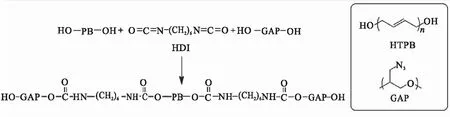
图25 HDI扩链剂制备HTPB-GAP嵌段聚合物Fig.25 Preparation of HTPB-GAP block copolymers from HDI chain extender
针对阳离子开环聚合制备HTPB-GAP嵌段聚合物反应时间长,而扩链剂方法容易凝胶,产物结构不可控等问题,Cappello等[56]提出了新的合成路线,即先将HTPB的羟基磺酰化,再与GAP反应生成GAP-HTPB-GAP嵌段聚合物,制备路线如图26所示。该反应过程简单迅速,且在较低温度下进行,副反应较少。制备的嵌段聚合物具有良好的稳定性,不易发生相分离。
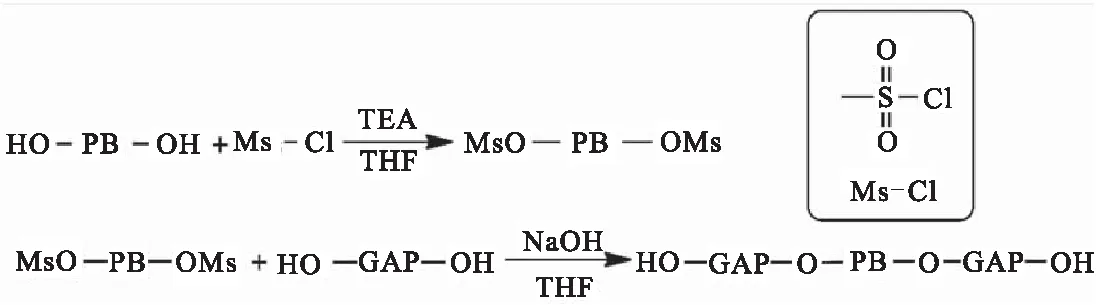
图26 HTPB磺酰化制备GAP-HTPB-GAP嵌段聚合物Fig.26 Preparation of GAP-HTPB-GAP block polymer by sulfonylation
为提高HTPB粘合剂极性,增强粘合剂与其他组分的相容性,柴春鹏等[57]研究了以HTPB为引发剂,在辛酸亚锡催化下,通过ε-已内酯开环聚合,制备聚已内酯-聚丁二烯-聚已内酯(PCL-PB-PCL)的三嵌段共聚物(图27)。研究表明,该共聚物中的两种组分有很好的相容性,共聚物的热稳定性随己内酯单体含量的增加而提高。Singh等[58]采用相同的方法制备了PCL-PB-PCL嵌段共聚物(HTBCP),以提高粘合剂与含能增塑剂NG的相容性能。研究表明,HTBCP不仅与HTPB性能相近,且与NG具有较好的相容性。与传统HTPB/DOA推进剂相比,HTBCP/NG推进剂的密度增加4.4%~5%,热值增加12%~15%,在7 MPa压力下燃烧速率提高了40%~70%。

图27 PCL-PB-PCL嵌段聚合物的制备Fig.27 Preparation of PCL-PB-PCL block copolymers
聚四氢呋喃主链中有大量的极性醚键,且低温性能优良,可与 HTPB反应形成嵌段共聚物,提高粘合剂综合性能。张万斌等[59-60]以HTPB为引发剂,在BF3·OEt2催化作用下,利用HTPB的羟基引发四氢呋喃及环氧丙烷开环聚合,制备出了一种具有全新结构的三嵌段共聚物粘合剂P(THF-co-PO)-b-PB-b-P(THF-co-PO),其分子由中间的聚丁二烯(PB)链段及两端的四氢呋喃-环氧丙烷共聚醚[P(THF-co-PO)]链段组成,聚合机理如图28所示。结果表明,合成的三嵌段共聚物具有优异的低温性能(Tg=-81.7 ℃)及与HTPB相似的热稳定性。

图28 HTPB存在条件下THF单体与PO单体阳离子开环共聚机理Fig.28 The mechanism for the ring-opening copolymerization of THF with PO in the presence of HTPB
为克服传统固体推进剂制备过程中异氰酸酯固化体系对环境湿度敏感的缺点,李娜等[61]以HTPB为引发剂,在BF3·OEt2催化作用下引发环氧氯丙烷发生阳离子开环聚合反应,形成两端为氯化聚醚的聚丁二烯(CTPB)。在碱性条件下,CTPB发生闭环反应形成端环氧基聚丁二烯(ETPB,图29),并考察了不同固化剂完全固化ETPB所需条件。结果表明,聚酰胺650是ETPB的合适固化剂,只需50 ℃下反应4 d就可完全固化,固化后的样品拉伸强度为4.5 ~5.1 MPa,断裂伸长率为150%~180%。
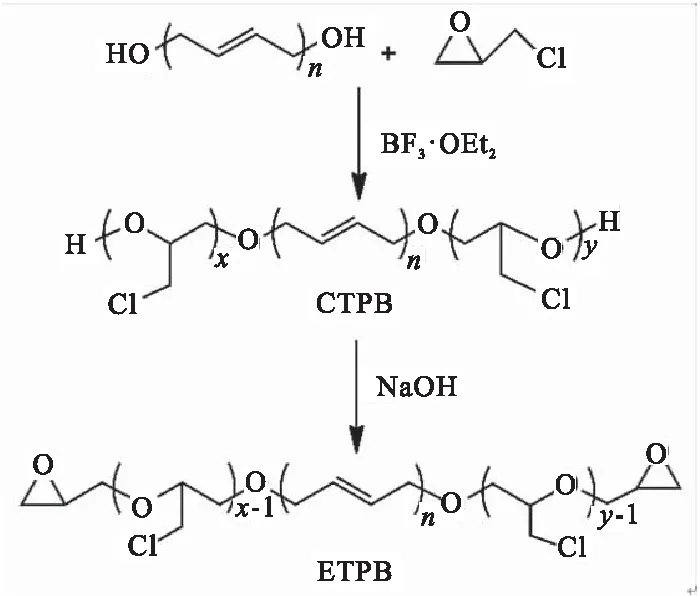
图29 ETPB的制备Fig.29 Preparation of ETPB
HTPB的嵌段共聚改性最大限度地保留了HTPB的主链结构。因此,可在保留HTPB原有优异性能的同时,赋予HTPB更好的综合性能,具有较好的应用前景。
5 问题与建议
与HTPB相比,经过化学改性后的HTPB分子极性、能量等性能得到提升,但各改性方法及产物也存在某些缺陷,主要体现在:
(1)端羟基改性反应过程容易实现,但取代羟基无疑会导致现有推进剂的异氰酸酯固化体系发生改变。基于HTPB的三唑交联固化体系对水不敏感,反应条件温和,但存在力学性能可调范围不大,交联网络结构不完善等问题,且分子中引入的叠氮基可与双键反应,发生自交联现象,影响贮存稳定性[62-63]。
(2)末端碳原子改性保留了末端羟基,对固化体系影响较小,且没有改变HTPB的微观结构。因此,改性后依然保留原HTPB的优良理化性能,但目前该方法反应路径及修饰基团选择较少。
(3)HTPB双键改性,特别是环氧化改性,具有难度较低、程度较高且可控的优点,生成的环氧基团可作为新的反应中心进一步改性,但双键的破坏及其他功能基团的引入,导致HTPB分子结构发生变化,进而引起粘合剂低温流动性能下降。
(4)嵌段改性保留了HTPB的主链结构,并赋予其更好的综合性能,但含有极性基团的均聚物大都具有较强的结晶趋向,不利于材料的热固化加工[64]。
(5)部分改性研究工作仍处于实验室研究阶段,离工程化应用尚有一定距离[65-66]。
为此,针对HTPB改性及应用,提出以下几点建议:
(1)在高分子物理学理论基础上,对HTPB的化学改性进行分子结构设计,进一步提高改性产物应用性能。
(2)通过优化反应工艺及路线,解决部分改性反应条件苛刻、周期长、成本高等问题,满足改性HTPB工程化应用需求。
(3)末端碳原子及嵌段共聚改性方法对HTPB分子结构影响较小,但针对末端碳原子改性的研究还比较少,需要进一步深入研究,拓展其应用领域。而嵌段改性在分子设计过程中,则应注意对嵌段共聚单体进行筛选,降低其结晶趋向,提高热稳定性。
(4)HTPB环氧化改性难度较低,改性程度可控,生成的环氧基团可作为新的反应中心进一步改性,因此具有重要的工程应用价值。同时,根据丁羟推进剂配方研制的实际情况,向HTPB分子结构中同时引入多种功能性基团,可赋予HTPB更多的功能特性(如界面键合、交联增强、燃速调节等)。
(5)在改性HTPB使用过程中,可将改性HTPB视作一种“聚合物功能助剂”,采用改性HTPB/HTPB共混使用的方式,通过控制添加量调节粘合剂基体的综合性能。这样既能保留HTPB粘合剂优良性能,又可进一步拓展改性HTPB的应用范围。
6 结束语
丁羟推进剂经历了数十年发展历程,已经具备了相当的研究积累。在此基础上,对HTPB粘合剂进行功能性改性,以满足更高的设计要求,是丁羟推进剂的发展趋势。从整体上来看,HTPB改性研究主要朝着以下几个方向在发展:
(1)向粘合剂分子中引入含能基团,如叠氮基、硝酸酯基、硝基等,提高丁羟推进剂的能量水平;
(2)通过改性提高HTPB的分子极性,如将聚四氢呋喃键合在HTPB分子两端形成嵌段共聚物,赋予共聚物大分子主链具有极性的特点,增强与其他组分间的相容性;
(3)将HTPB端羟基修饰成为叠氮基、炔基以及环氧基团,利用点击化学、环氧-胺固化体系取代传统聚氨酯固化体系,以避免环境水分对固化过程的影响;
(4)小分子功能助剂,如二茂铁燃烧催化剂接枝在HTPB分子主链上,制备新型聚合物型助剂,以解决催化剂迁移问题。
目前,HTPB改性技术仍处于一个发展的平台区,改性产品应用于具体型号当中的案例还比较少,不同的改性方法也各有优劣。在实际过程中,还是要根据丁羟推进剂配方研制的实际情况与需求,在分子设计基础上,选取合适的改性路径,在尽可能保留HTPB现有性能基础上,进一步提高其综合性能。同时,应开展各学科、各专业的交叉融合,推进剂HTPB改性技术的不断创新。
