Aurora-B激酶抑制剂AZD1152-HQPA的合成*
2020-05-12王丽丽王兴兰马才玉毛远湖张吉泉
王丽丽,王兴兰,马才玉,程 飞,毛远湖,汤 磊,张吉泉
(1 贵州医科大学医药卫生管理学院,贵州 贵阳 550025;2 贵州医科大学药学院,贵州 贵阳 550025)
抗肿瘤药物是创新药物研究的重要领域。传统的抗肿瘤药物存在选择性低、副作用大等缺点,以细胞信号传导通路中关键信号蛋白为靶点的小分子激酶抑制剂的研究成为近年来抗肿瘤药物研究的热点,通过选择性抑制肿瘤细胞赖以生存和增殖的关键蛋白分子,起到靶向抗肿瘤作用。
Aurora激酶家族是1995年在真核生物中发现的丝氨酸/苏氨酸激酶家族,哺乳动物细胞表达Aurora-A、Aurora-B及Aurora-C三类亚型。Aurora激酶在细胞内的表达时间和空间上都受到严格的控制,其异常表达与肿瘤细胞存活、增殖、转移及死亡等过程密切相关[1-2]。Aurora激酶抑制剂的开发与研究越来越受到人们的重视。现今已报道的Aurora激酶抑制剂按照骨架结构可分为喹唑啉类、喹啉类、嘧啶类及咪唑并吡啶等类型[3]。AZD1152是AstraZeneca公司开发的第一个临床上可用的Aurora-B选择性激酶抑制剂,属喹唑啉类化合物(图1)。据报道,AZD1152已进入临床II期试验[4],目前未见进一步的临床研究报道,围绕该化合物开展的相关机制研究仍持续引起研究者的兴趣。
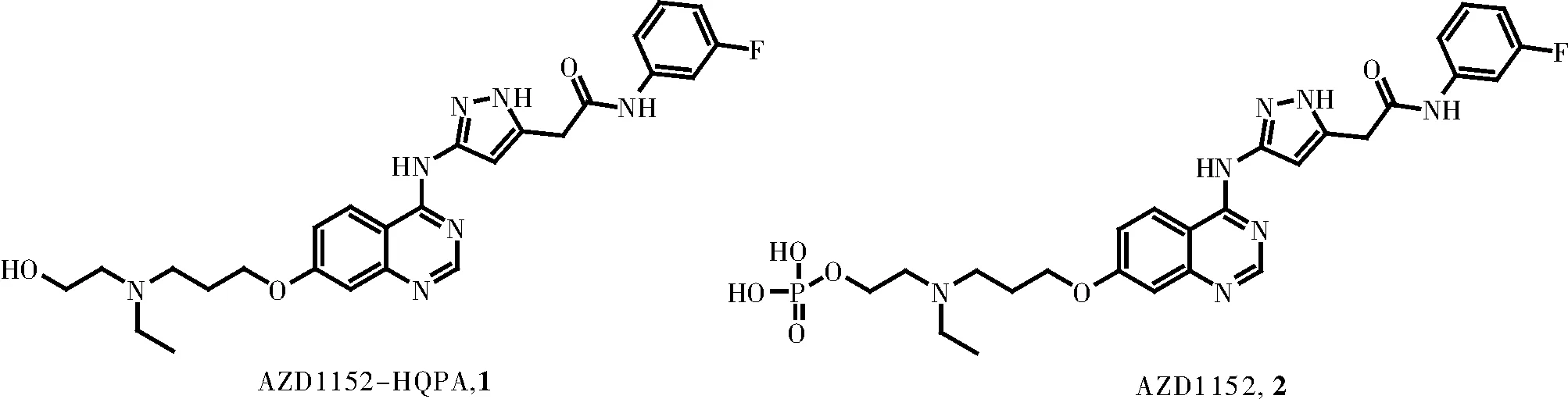
图1 AZD1152及AZD1152-HQPA的结构
AZD1152是一个磷酸酯类前药,其前体为AZD1152-HQPA(图1)。现有文献[2,5-8]对AZD1152-HQPA的合成多以2-氨基-4-氟苯甲酸为起始原料,经环合、取代、氯代、氨化、酰胺化以及最后取代等六步转化反应得到(图2)。该路线反应常规、操作简便,且大部分原料便宜易得,可用于AZD1152-HQPA的放大合成。本研究通过对文献报道路线进行对比分析,发现在中间体8与间氟苯胺缩合制备9时,使用到价格昂贵的羧基活化试剂三氟乙酸无氟苯酯(TFA-OPFP),且用量为2.0当量,不利于整条路线的成本控制。本研究重点对该步反应做了优化,发现使用价格便宜的缩合剂苯并三氮唑-N,N,N′,N′-四甲基脲六氟磷酸盐(HBTU)在碱性DIPEA条件下,可以86.30%收率得到缩合产物,且反应体系较为干净,便于后处理纯化,可较大地降低生产成本。此外,本研究也对中间体5的氯代反应进行了优化,通过缩短反应时间可显著减少副产物的生成,提高反应收率。
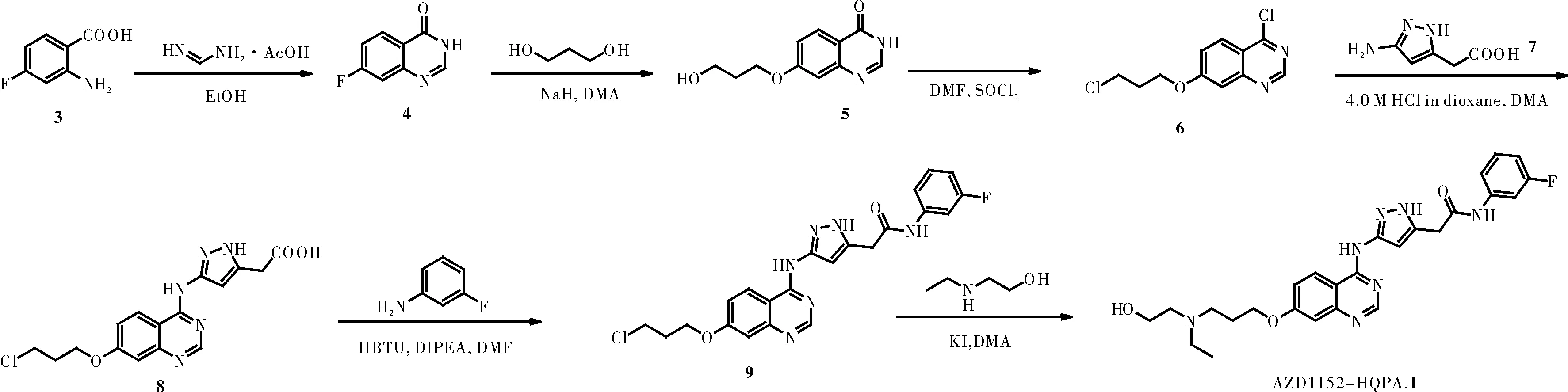
图2 AZD1152-HQPA的合成路线
1 实 验
1.1 主要仪器与试剂
Bruker Avance NEO 600 MHz核磁共振波谱仪,德国Bruker公司;Xevo G2-XS QTof 高分辨质谱仪,美国Waters公司;Thermo UltiMate 3000型UPLC-TSQ Quantum型三重四级杆串联质谱,美国Thermo Finnigan公司;Nexera-i LC-2040C液相色谱仪,日本SHIMADZU公司;色谱柱(Welchrom-C18);ZF-2 型三用紫外仪,上海安亭仪器厂;WRX-4型显微熔点仪,上海易测仪器设备有限公司。
原料和试剂均为市售,如无特别说明,均直接使用。2-(3-氨基-1H-吡唑-5-基)乙酸(中间体7)参考文献[2,8]合成。
1.2 7-氟-喹唑啉-4(3H)-酮(4)的合成[9]
于500 mL圆底瓶中加入4-氟-2-氨基-苯甲酸(20.00 g,0.13 mol)及EtOH(350 mL),搅拌下加入醋酸甲脒(17.45 g,0.17 mol),加热至回流反应24 h。TLC监测反应完后,减压蒸去溶剂适量,将反应液倒入冰水中。过滤,滤饼用50%乙醇淋洗,收集滤饼并于50 ℃减压真空干燥12 h,得到白色固体产物18.23 g,产率85.50%。ESI-MS:m/z165.1 [M+H]+。产物直接用于下步反应。
1.3 7-(3-羟基丙基)喹唑啉-4(3H)-酮(5)的合成[2]
于250 mL三口瓶中加入1.3-丙二醇(13.91,0.18 mol)及DMA(20.00 mL),冷却至0 ℃,搅拌下分批加入60% NaH(7.32 g,0.18 mol),待反应体系稳定后分批加入中间体4(5.00 g,0.03 mol),随后缓慢加热至60 ℃反应10 min,然后加热至110 ℃反应3 h。TLC监测反应完全后,0 ℃下加H2O(300 mL)淬灭反应,0.1 M HCl调节pH至6左右,析出白色固体,抽滤,滤饼用H2O(100 mL)淋洗,收集固体于50 ℃下减压真空干燥12 h,得到白色固体产物6.42 g,产率95.50%。ESI-MS:m/z221.1[M+H]+。产物直接用于下步反应。
1.4 4-氯-7-(3-氯丙基)喹唑啉-4(3H)-酮(6)的合成[10]
取中间体5(5.00 g,22.72 mmol)混悬于SOCl2(50.00 mL)中,加入干燥DMF(1.00 mL),加热至85 ℃回流反应1.0 h。将反应液冷却至40~50 ℃,减压蒸馏除去剩余的SOCl2,向残留物中加入甲苯(5.0 mL×2)带走残留SOCl2。加入CH2Cl2(100 mL)溶解残留物,依次用饱和NaHCO3溶液(50 mL×3)、饱和NaCl溶液(50 mL)洗涤,有机相用MgSO4干燥。减压蒸去溶剂,将粗产物混悬于甲基叔丁基醚(30.00 mL)中,搅拌30 min。抽滤,收集滤液并减压蒸去溶剂,得类白色固体4.93 g,收率84.70%。ESI-MS:m/z257.1[M+H]+。产物直接用于下步反应。
1.5 2-(3-((7-(3-氯丙基)喹唑啉-4-)氨基)-1H-吡唑-5-基)乙酸(8)的合成[2]
于50 mL反应瓶中加入中间体6(4.00 g,15.62 mmol)及2-(3-氨基-1H-吡唑-5-基)乙酸(7,2.20 g,15.62 mmol),加入DMA(20.00 mL)溶解,随后加入4.0 M HCl的二氧六环溶液(2.00 mL),加热至90 ℃反应3 h。反应液冷却至室温,将反应液倒入H2O(300.00 mL)水中,析出红棕色固体。抽滤,收集滤液并过硅藻土后用1.0 M NaOH溶液调pH至5~6时,析出固体。抽滤,滤饼于50 ℃下减压真空干燥12 h得4.62 g浅棕色固体,产率82.00%。ESI-MS:m/z362.1[M+H]+。产物未经纯化直接用于下步反应。
1.6 2-(3-((7-(3-氯丙基)喹唑啉-4-)氨基)-1H-吡唑-5-基)-N-(3-氟苯基)乙酰胺的合成(9)
于50 mL反应瓶中依次加入中间体8(3.00 g,8.31 mmol)、HBTU(3.78 g,9.97 mmol)及DMF(10 mL),搅拌溶解后加入DIPEA(2.06 mL,12.46 mmol),室温搅拌0.5 h。随后,加入间氟苯胺(1.60 mL,16.62 mmol),加热至90 ℃反应12 h。将反应体系倒入0.1 M HCl(100.00 mL)冰水浴中,析出固体,抽滤,滤饼依次用H2O(100.00 mL)及甲基叔丁基醚(100.00 mL)淋洗后干燥得棕色固体3.26 g,产率86.30%。ESI-MS:m/z455.2[M+H]+。产物未经进一步纯化直接用于下步反应。
1.7 2-(3-((7-(3-(乙基(2-羟基乙基)氨基)丙基)喹唑啉-4-)氨基)-1H-吡唑-5-基)-N-(3-氟苯基)乙酰胺的合成(AZD1152-HQPA,1)
取中间体9(2.50 g,5.50 mmol)溶于DMA(8.0 mL)中,搅拌下依次加入N-乙基乙醇胺(1.96 g,22.00 mmol)及KI(1.83 g,11.00 mmol),氮气保护下加热至80 ℃反应24 h。反应体系冷却至室温,直接用硅胶柱层析(VCH2Cl2:VCH3OH:VNH3·H2O=10:1:0.8)分离得白色固体1.58 g,即目标化合物AZD1152-HQPA,收率56.5%。纯度:99.2%;1H NMR (600 MHz, DMSO-d6) δ 12.39 (s, 1H), 10.45 (s, 1H), 10.23 (s, 1H), 8.52 (s, 2H), 7.62~7.64 (m, 1H), 7.32~7.38 (m, 2H), 7.14~7.17 (m, 2H), 6.88~6.91 (m, 1H), 6.75 (d,J=1.0 Hz, 1H), 4.32 (d,J=6.3 Hz, 1H), 4.17 (t,J=6.3 Hz, 2H), 3.75 (s, 2H), 3.51 (t,J=5.6 Hz, 1H), 3.44 (t,J=6.6 Hz, 2H), 2.69~2.73 (m, 1H), 2.59 (t,J=6.9 Hz, 2H), 1.88 (p,J=6.5 Hz, 2H), 1.07 (t,J=7.2 Hz, 1H), 0.96 (t,J=7.1 Hz, 3H);13C NMR (150 MHz, DMSO-d6) δ 163.41, 162.51, 161.81, 155.57, 141.35, 141.28, 130.92, 130.86, 125.20, 118.08, 115.31, 110.31, 110.17, 109.46, 107.93, 106.45, 106.27, 66.66, 59.82, 56.14, 50.27, 48.16, 43.35, 27.04, 14.02, 12.37. HRMS-(ESI)(m/z): [M+H]+calcd for C26H31FN7O3: 508.2484; found: 508.2472。
2 结 论
本以2-氨基-4-氟苯甲酸为起始原料,经环合、取代、氯代、氨化、酰胺化以及最后取代等六步反应得到目标化合物AZD1152-HQPA,总收率27.6%,产物纯度大于99%。目标化合物结构经高分辨质谱、核磁共振氢谱及碳谱确证。该路线原料便宜易得、反应操作简便,适合AZD1152-HQPA的放大制备。
本研究路线第三步氯代反应,通过条件摸索发现反应时间对收率的影响至关重要,随着反应时间的延长(大于1.0 h),副产物增多,收率降低。从中间体8合成9的酰胺化反应是本研究优化的重点,文献报道使用2.0当量的TFA-OPFP作为缩合剂,该试剂价格昂贵,且大量使用存在环境污染。本研究先后探索了(COCl)2/DIPEA、DCC/DMAP、HATU/DIPEA、HBTU/DIPEA、SOCl2/TEA等常用缩合反应体系,最终发现使用HBTU/DIPEA作为缩合条件,反应收率较高,且体系较为干净,便于后处理纯化。
