基于微滤法处理水样中226Ra的α能谱分析技术
2020-05-07梁永广於国兵夏明明顾先宝
梁永广,王 元,2,於国兵,夏明明,顾先宝,范 然,陈 志,*
(1.中国科学技术大学 物理学院,安徽 合肥 230022;2.郑州大学附属肿瘤医院,河南 郑州 450008;3.安徽省辐射环境监督站,安徽 合肥 230071)
Ra作为碱土族金属,与元素Ca、Sr、Ba有相似的化学性质,易通过食物链富集等方式进入人体[1],并在骨骼中富集,对人体进行持续的内照射,增加致癌风险[2]。自然界天然存在的Ra的同位素有223Ra(T1/2=11.4 d)、224Ra(T1/2=3.6 d)、226Ra(T1/2=1 600 a)以及228Ra(T1/2=5.75 a),223Ra、224Ra及226Ra都属于α放射性核素,228Ra属于β放射性核素[3-4]。其中226Ra半衰期最长,发射的α粒子能量为4.60 MeV(6.16%)和4.78 MeV(93.84%),属于极毒组放射性核素[5]。226Ra的母体238U在地壳中含量相对较高,因此自然界中尤其是铀矿周边的226Ra相对富集[6]。磷酸盐工业、伴生矿的开采以及矿石的选冶加工等人类活动会增加水体中226Ra的含量[7]。根据欧洲原子能共同体的标准,饮用水中226Ra的监管限值为40 mBq/L[8],因此从辐射监管、辐射剂量估算的角度来看,准确测量水样品中226Ra的含量有非常重要的意义。
在我国,目前测量水中226Ra的方法主要是氡射气法和γ能谱法。氡射气法已被很多实验室广泛使用,测量技术相对成熟。但该方法存在一些不足[9]:1) 低活度样品的测量需要较大的样品量,如环境中低活度的水样品一般需要5~20 L,增加了样品的富集时间;2) 测量的是氡子体,等待氡与衰变子体平衡的时间较长,一般需封存20 d才可测量,不利于事故情况下的应急监测;3) 分析测量过程中,没有考虑226Ra的实际回收率;4) 样品计数低,普通环境样品一般为十几到几十个计数,测量不确定度大,且易受外界环境的影响。γ能谱测量技术相对成熟、操作较简单,也是很多实验室的主要选择,普遍的做法是通过测量226Ra的衰变子体214Pb、214Bi发射的特征γ射线来间接测定226Ra的活度[10]。γ能谱法的不足在于[10]:1) 方法探测限较高,一般方法的探测限为0.08 Bq/m3 [11],低水平样品测量需要较大的样品量,样品处理难度较大;2)226Ra的衰变子体生长平衡时间较长,约20 d。由于实际监测的需要,研究快速且准确的测量方法很有必要。
α能谱法可直接测量226Ra发射的α粒子,无需等待226Ra-222Rn达到放射性平衡,适于事故情况下的应急监测。另外,α能谱仪能测量较低的本底水平,且探测限也较低,更加适用于低活度样品的测量分析[12]。基于此,该方法受到越来越多的关注[13-14]。电沉积方法可制备高分辨率的α能谱样品,但Ra在电沉积过程中易受其他金属阳离子(如Fe3+等)的干扰,对样品分离纯化要求较高[15];且电沉积制样中电化学过程会使电沉积液温度升高,导致溶液体积蒸发而减少,影响实际回收率[16];此外电沉积制样所用设备需耐酸碱腐蚀,阳极丝材料为铂金,通常价格较高。故本研究拟采用真空微滤Ba(Ra)SO4共沉淀的方法,以有效避免绝大多数金属阳离子的干扰,制得满足测量要求的α能谱样品。
1 方法
1.1 仪器
PIPS型硅半导体α谱仪,ORTEC公司,能量分辨率优于20 keV(FWHM,5 485.56 keV214Am),本底水平低至9.167×10-5s-1;BT224S型精密电子天平,Sartorius公司,感量为0.1 mg;真空抽滤装置,AutoScience公司;TD5Z台式低速离心机,湖南凯达科学仪器有限公司。
1.2 试剂
14 mol/L浓硫酸,广东市东红化工厂;冰醋酸(纯度≥99.5%)、二水合乙二胺四乙酸二钠(C10H14N2Na2O8·2H2O)、浓氨水(25%~28%)、乙醇(≥95%),Aladdin公司;Ba2+标准溶液(100 μg/mL)、Pb2+溶液(10 mg/mL),国家有色金属及电子材料分析测试中心;饱和硫酸钾溶液(25 ℃),由Aladdin公司的硫酸钾固体(纯度99%)配制得到。除特殊说明外所使用的试剂均为分析纯,实验用水为去离子水。
228Th-224Ra标准溶液(活度浓度为0.300 Bq/mL)、226Ra标准溶液(活度浓度为0.417 Bq/mL),核工业北京地质研究院。
1.3 样品制备
1) 取一定量的228Th-224Ra标准溶液以及适量的226Ra标准溶液加入到50 mL去离子水中,搅拌均匀。随后加入2 mL浓硫酸进行酸化,搅拌均匀。
2) 向烧杯内加入100 mg Pb载体,并加入过量的饱和硫酸钾溶液,充分搅拌,待生成Pb(Ra)SO4沉淀后,静置2 h。
3) 离心分离出Pb(Ra)SO4沉淀,加入50 mL去离子水洗涤沉淀。
4) 用3 mL氨水溶解1 g EDTA-2Na固体,随后将其加入到洗涤后的Pb(Ra)SO4沉淀中,搅拌以络合溶解沉淀。
5) 加入70 μg(以Ba2+计)Ba2+以及过量饱和硫酸钾溶液,并用冰醋酸调节溶液pH值至约4.5。
6) 用真空微滤装置将沉淀物抽滤到孔径0.1 μm、直径25 mm的纤维素微孔滤膜上,微滤瓶内的相对压强为-0.08 MPa,微滤样品的有效活性区直径为21 mm。考虑到冰醋酸的凝固点较低,用50 ℃温水多次洗涤滤膜表面沉淀。微滤共沉淀样品制备完成后,在真空干燥箱内烘干,随后用α谱仪测量适当时间。
1.4 示踪剂224Ra及其子体
226Ra的同位素中225Ra为最适宜的示踪剂[17],但因225Ra并非天然存在,昂贵不易获得,故本实验选取224Ra作为示踪剂,用以确定整个过程中的回收率。根据二者特征α能峰面积之间的关系计算226Ra的活度。
224Ra和226Ra及其各自α衰变子体的半衰期、主要特征能量与分支比列于表1。由表1可知,224Ra与222Rn、210Po的特征能量接近,能峰易重叠,220Rn与212Bi、218Po的特征能量接近,能峰易重叠。而216Po发射的主要特征α粒子的能量则不与其他衰变子体相近,能峰易区分,不受其他核素的干扰。实际测量过程中,通过216Po的特征峰面积来确定示踪剂224Ra的特征峰面积,即:
NRa-224=kNPo-216
(1)
其中:NRa-224为224Ra的特征α能峰面积;NPo-216为216Po的特征α能峰面积;k为224Ra与216Po的特征α能峰面积的经验修正系数。
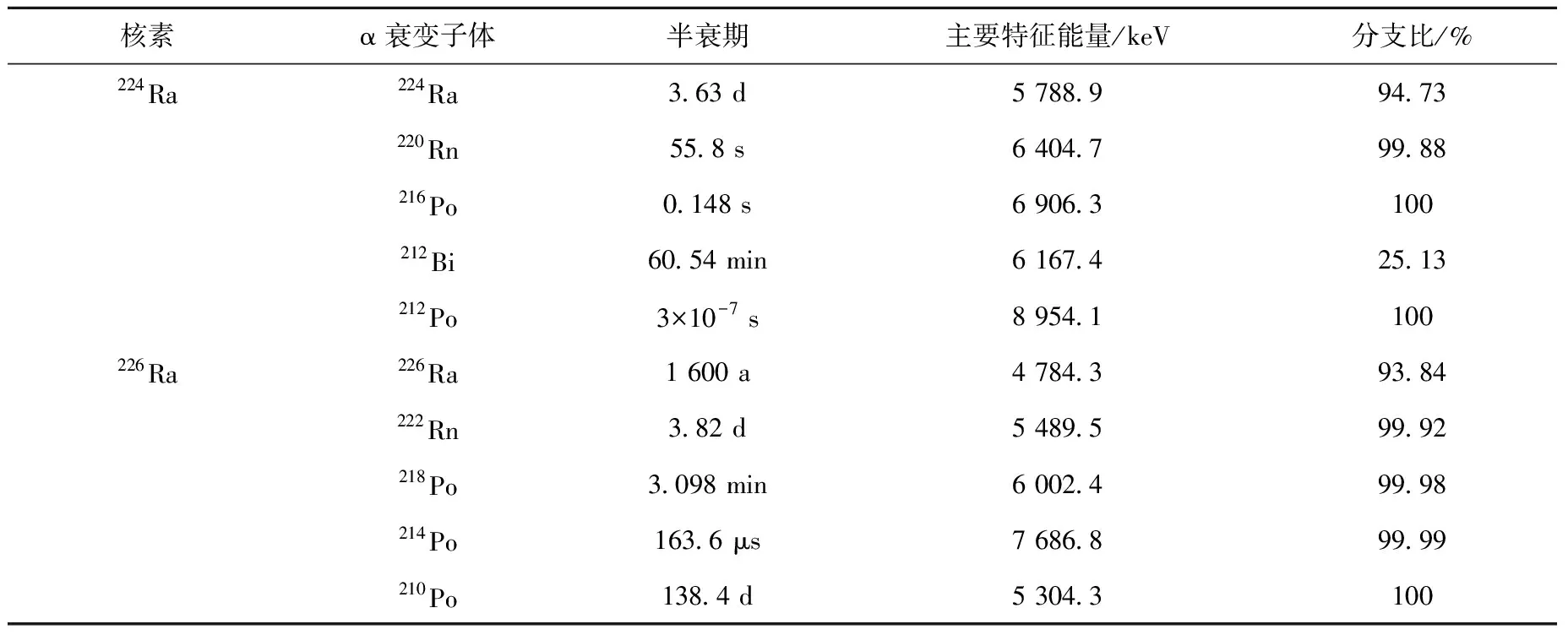
表1 224Ra和226Ra及其各自α衰变子体的半衰期、主要特征能量与分支比Table 1 Half-life, main characteristic energy and branch ratio of 224Ra and 226Ra and their respective α decay daughter
1.5 样品中226Ra活度的计算公式
224Ra的半衰期较短,需对其在制源及测量过程中进行衰变修正,包括放置时间修正和测量时间修正。
aRa-226=aRa-224e-λ(t1-t0)×
(2)
其中:aRa-226为待测样品中226Ra的活度,Bq;aRa-224为添加示踪剂224Ra的活度,Bq;NRa-226为226Ra的特征α能峰的峰面积;λ为224Ra的衰变常量;t0为Pb(Ra)SO4开始沉淀的时刻;t1为α谱仪测量开始时刻;t2为α谱仪测量结束时刻。
2 结果与讨论
2.1 226Ra及其子体峰面积随时间的变化


图1 226Ra加标实验获得的α能谱Fig.1 α spectrum obtained by measuring 226Ra spiked experiment
将所制微滤源样品放置1 d,再次进行测量,结果示于图2。由图2可见,226Ra衰变子体的峰面积有明显增大。

图2 1 d后再次测量微滤源获得的α能谱Fig.2 α spectrum obtained by measuring micro-filtration source again after 1 d
继续放置若干天,重复测量所制微滤源,得到的226Ra衰变子体的α能峰面积随时间的变化示于图3。
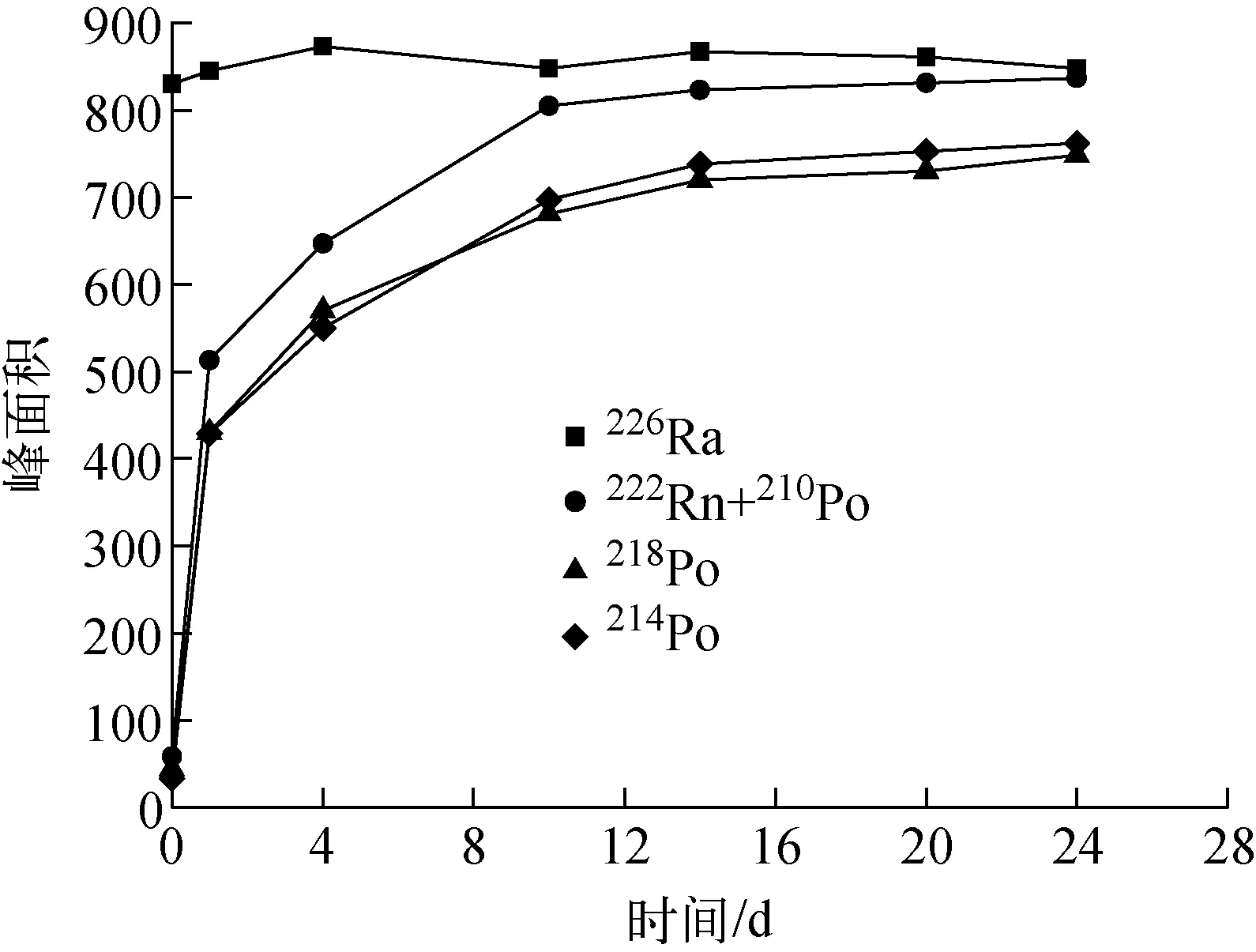
图3 226Ra衰变子体的α能峰面积随时间的变化Fig.3 α spectrum peak area of decay daughter of 226Ra vs. time
226Ra因其半衰期较长,峰面积在整个测量过程中基本保持不变。226Ra衰变子体的峰面积与时间近似呈指数关系,且经过15 d后,各子体的峰面积基本保持不变,近似与226Ra峰面积相等。其中,218Po与214Po的峰面积略低于226Ra峰面积,是因为微滤源未进行封源处理,在测量过程中有部分222Rn逸出。
2.2 224Ra及其子体峰面积之间的关系
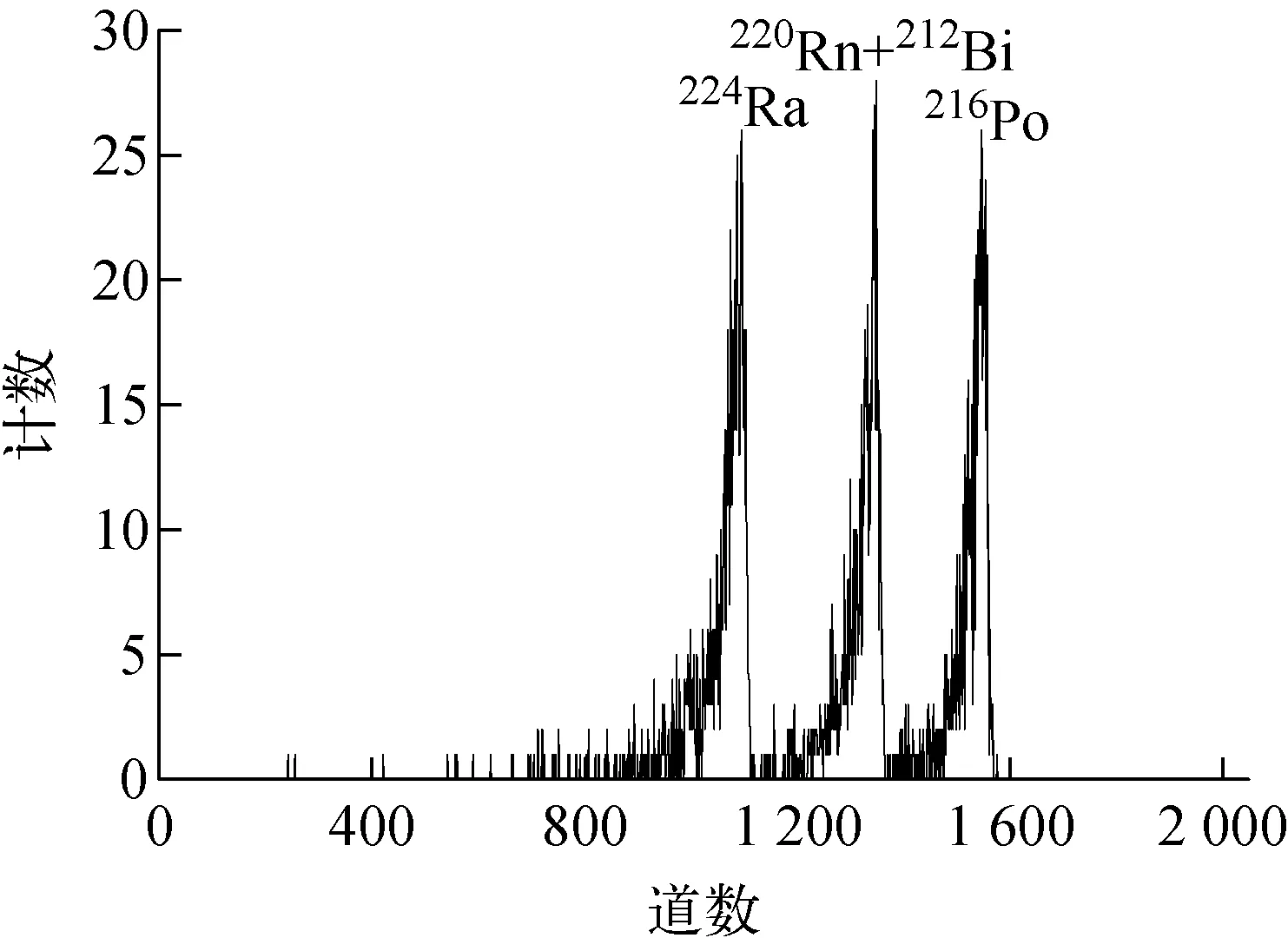
图4 224Ra加标实验获得的α能谱Fig.4 α spectrum obtained by measuring 224Ra spiked experiment
将224Ra标准溶液通过上述Pb(Ra)SO4共沉淀的方法制得微滤源,并在α谱仪上测量,结果示于图4。由于220Rn半衰期为55.8 s、216Po的半衰期为0.148 s,均非常短,理论上3种核素之间在7 min就能达到放射性平衡。但由于220Rn的析出损失,造成实验所获得的3种核素的特征能量峰面积并不相等。
通过微滤样品制备方式得到15个平行样品,用α谱仪分别测量,得到了224Ra峰面积与216Po峰面积的经验修正系数k,如表2所列。可见以224Ra作为示踪剂时,k约为1.14。
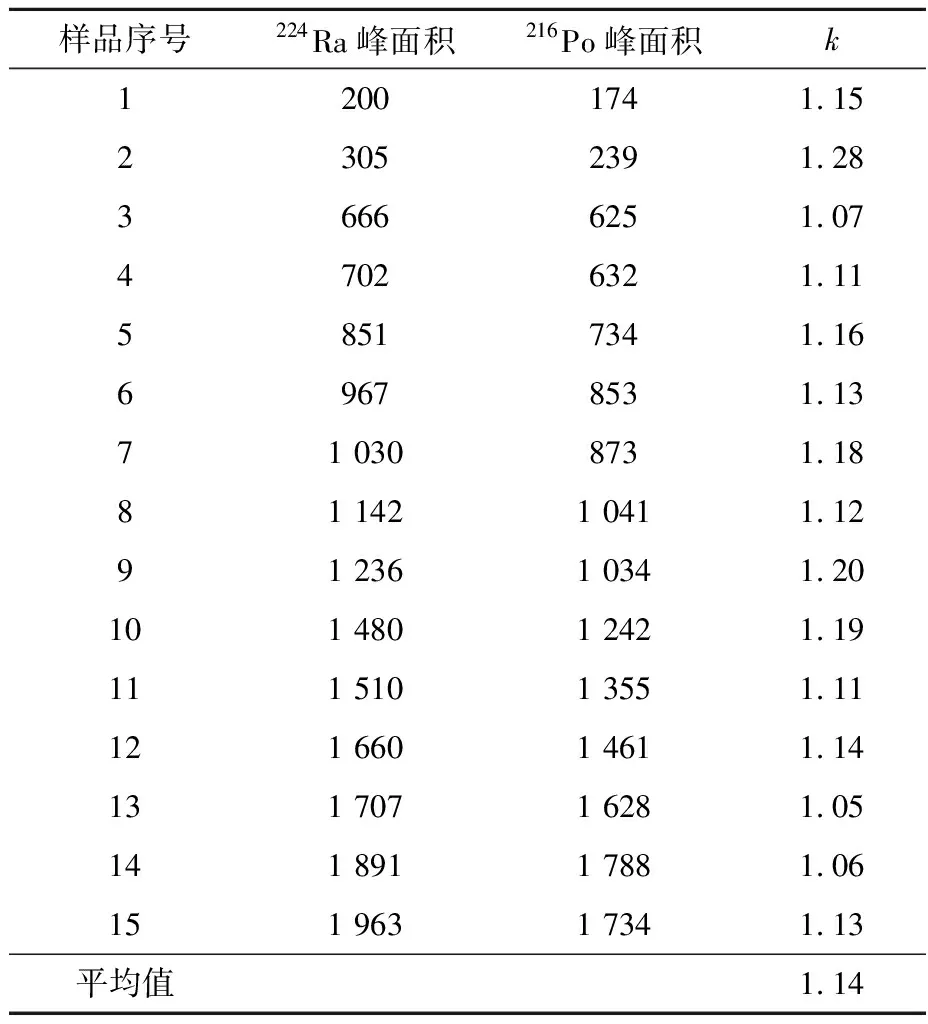
表2 224Ra作为示踪剂时224Ra与216Po峰面积的关系Table 2 Relationship between peak areas of 224Ra and 216Po at 224Ra as tracer
2.3 226Ra加标样品测量结果
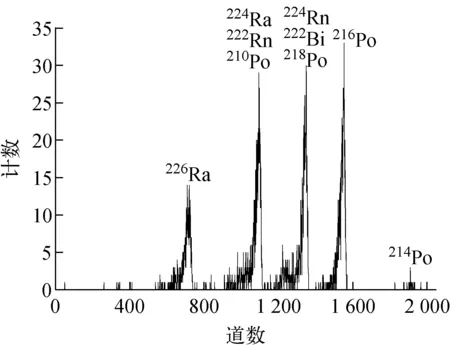
图5 测量加标样品中224Ra所获得的α能谱Fig.5 α spectrum obtained by measuring spiked sample with 224Ra as tracer
以224Ra作为示踪剂,分别测量226Ra含量为33.36、62.55、104.25、208.5、417 mBq的加标样品,结果示于图5。根据式(1)、(2)计算226Ra的活度,计算结果列于表3。
由表3可见,测量结果与实际添加量的相对偏差为0.4%~13.1%,说明了以224Ra为示踪剂的α能谱微滤法测量226Ra的准确性以及可靠性。
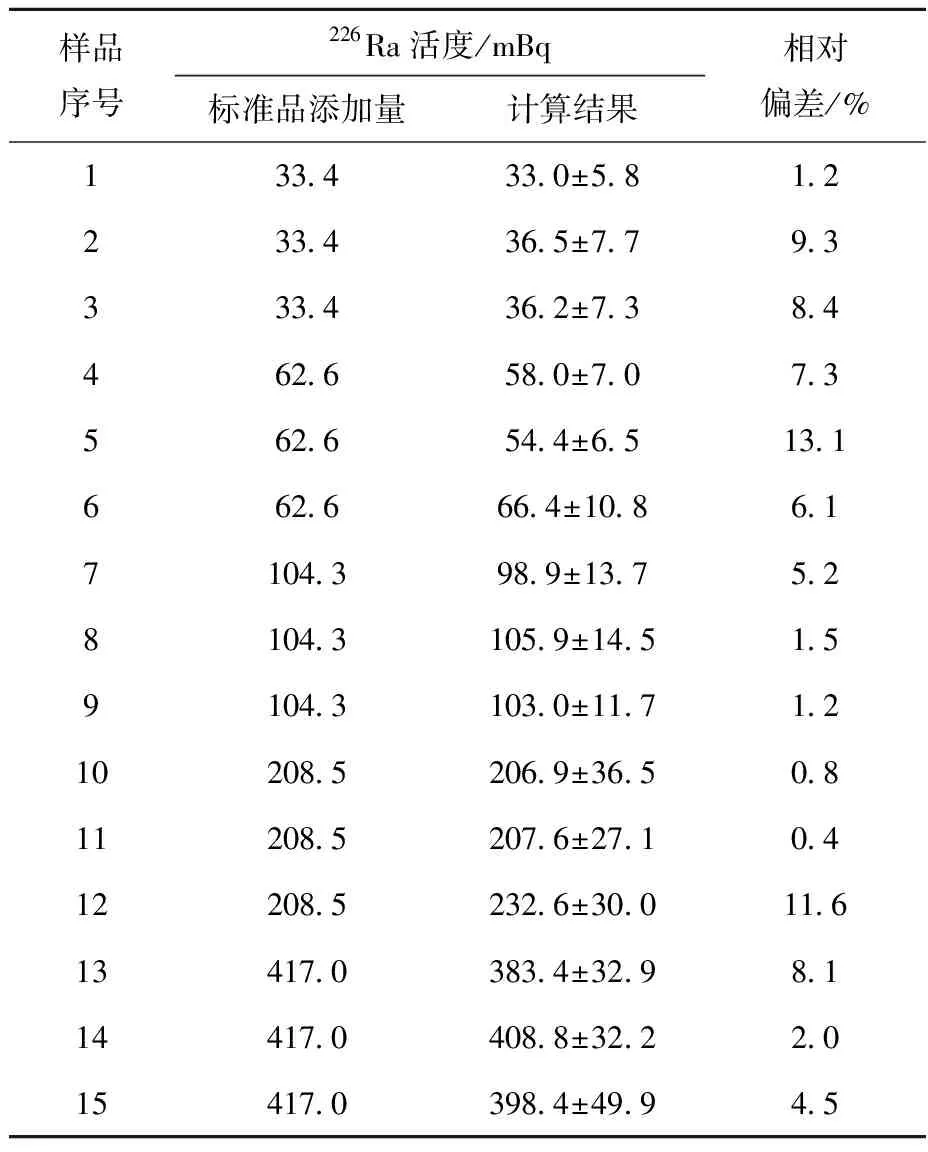
表3 加标样品测量结果Table 3 Measurements of spiked sample
注:测量不确定度时k取2
基于《水中镭-226的分析测定》(GB 11214—1989)[18],对上述5个活度梯度(33.4~417.0 mBq)的同一批样品采用氡射气法进行测量,具体方法为:取50 mL待测样品于烧杯中,加入2 mL Fe-Ca载体混合溶液,充分搅拌均匀后加入10 mL饱和Na2CO3溶液,调节溶液pH值为9~10,静置澄清后,虹吸去除上层清液,加入盐酸溶解沉淀,在电加热板上蒸发减容至30 mL,转移至扩散器中,密封15 d,在室内氡钍分析仪上测量3~5 h,结果列于表4。由表4可见,226Ra的测量值与实际值的相对偏差为6.0%~9.3%。
本研究所采用的基于微滤法的α能谱分析的测量结果与采用GB 11214—1989方法的测量结果相比一致性较好,满足测量要求。但本研究的制样、测量时间仅需2~3 d,相对于氡射气法,大幅缩短了样品分析时间,具有一定的优越性。
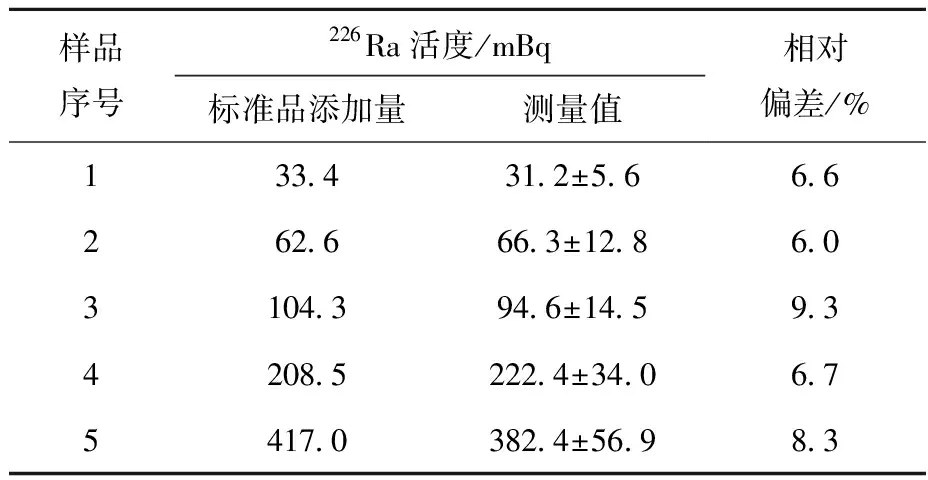
表4 加标样品氡射气法测量结果Table 4 Measurements of spiked sample via radiance method
注:测量不确定度时k取2
2.4 测量方法的最小可探测活度
在95%的置信水平下,最小可探测活度(MDA)可通过式(3)进行计算:
(3)
式中:tback为本底情况下的测量时间;b为感兴趣区的本底计数率,s-1;η为计数效率;P为所测核素发射α粒子的分支比,对于226Ra,P=100%。根据实际测量,b=2、η=14.53%、tback=86 400 s,将上述数据代入式(3)计算得MDA=0.74 mBq。
3 结论
1) 通过Pb(Ra)SO4载带预富集水中的Ra,再利用Ba(Ra)SO4共沉淀并将沉淀物抽滤到微孔滤膜上,制成了可直接用于α谱测量的微滤样品,整个样品制备过程简单,α谱测量快速准确,MDA达0.74 mBq,远小于γ能谱法测量的探测限0.08 Bq/m3。
2) 使用天然224Ra作为示踪剂,通过实验得到了216Po与224Ra能峰面积之间的关系,实现了对整个实验过程的示踪,解决了方法回收率和225Ra示踪剂不易获得的问题。
3) 在制备微滤源的过程中,若沉淀物不能均匀分布在微孔滤膜上,则微滤层厚度不均匀,部分区域自吸收严重,导致在α能谱测量过程中能峰拖尾严重,引起较大的偏差。
本研究为辐射监管部门快速测量水中低活度的226Ra提供了一种新的方法,改善了传统方法测量过程中的耗时长、缺少示踪剂等不足。
