冯了性风湿跌打药酒的HPLC指纹图谱建立及其中10种有效成分的含量测定
2020-05-06胡静杨媛媛崔小敏任慧雷琨陈志永
胡静 杨媛媛 崔小敏 任慧 雷琨 陈志永
摘 要 目的:建立冯了性风湿跌打药酒的高效液相(HPLC)指纹图谱,并测定其中10种有效成分的含量。方法:采用HPLC 法。色谱柱为Inertsil ODS-3 C18,流动相为乙腈-0.1%磷酸水溶液(梯度洗脱),流速为1.0 mL/min,柱温为25 ℃,检测波长为210 nm(10~15 min,盐酸麻黄碱、盐酸伪麻黄碱)、300 nm(70~120 min,桂皮醛)、345 nm(15~70 min,新绿原酸、东莨菪苷、绿原酸、隐绿原酸、东莨菪内酯、异绿原酸A、异绿原酸C),进样量为10 μL。以东莨菪内酯峰为参照,绘制10批样品的HPLC指纹图谱,采用《中药色谱指纹图谱相似度评价系统(2012版)》进行相似度评价,确定共有峰。结果:10批样品共有18个共有峰,相似度均大于0.980,共指认出盐酸麻黄碱、盐酸伪麻黄碱、新绿原酸、东莨菪苷、绿原酸、隐绿原酸、东莨菪内酯、异绿原酸B、柚皮苷、异绿原酸A、异绿原酸C、桂皮醛等12个成分。盐酸麻黄碱、盐酸伪麻黄碱、新绿原酸、东莨菪苷、绿原酸、隐绿原酸、东莨菪内酯、异绿原酸A、异绿原酸C、桂皮醛等10个成分检测质量浓度的线性范围分别为2.475~247.5、2.600~260.0、3.820~382.0、3.900~390.0、3.060~306.0、4.760~476.0、4.540~454.0、4.900~490.0、4.540~454.0、7.590~759.0 μg/mL(r 均大于 0.999 0);定量限分別为0.320 0、0.350 0、0.021 4、0.024 3、0.036 8、0.025 7、0.152 1、0.042 9、0.025 1、0.350 3 μg/mL,检测限分别为0.160 0、0.180 0、0.007 9、0.004 0、0.001 2、0.007 3、0.076 0、0.001 4、0.008 1、0.201 4 μg/mL;精密度、重复性、稳定性试验的RSD均小于2%;加样回收率为95.03%~106.85%(RSD为0.67%~2.68%,n=6);上述10种成分的含量分别为0.013 3~0.214 1 mg/mL。结论:所建指纹图谱稳定、准确、专属性强,可用于冯了性风湿跌打药酒的质量控制;含量测定方法简便快速、准确可靠,可用于同时测定其中10种有效成分的含量。
关键词 冯了性风湿跌打药酒;高效液相色谱法;指纹图谱;含量测定
ABSTRACT OBJECTIVE: To establish an HPLC fingerprint for Fengliaoxing fengshi dieda wine, and to determine the contents of 10 effective constituents. METHODS: HPLC method was adopted. The determination was performed on Inertsil ODS-3 C18 column with mobile phase consisted of acetonitrile -0.1% phosphoric acid water (gradient elution) at the flow rate of 1.0 mL/min. The column temperature was 25 ℃, and detection wavelength was set at 210 nm(10-15 min, hydrochloride ephedrine, hydrochloride pseudoephedrine), 300 nm(70-120 min, cinnamaldehyde), 345 nm(15-70 min, neochlorogenic acid, scopolin, chlorogenic acid, cryptochlorogenic acid, scopoletin, isochlorogenic acid A, isochlorogenic acid C). The sample size was 10 μL. Using scopoletin peak as reference, HPLC fingerprints of 10 batches of samples were drawn. The similarity evaluation was performed by using Similarity Evaluation System of TCM Chromatographic Fingerprint (2012 edition) to confirm common peak. RESULTS: There were 18 common peaks in HPLC chromatograms of 10 batches of samples, and the similarity was above 0.980. Totally 12 components including hydrochloride ephedrine, hydrochloride pseudoephedrine, neochlorogenic acid, scopolin, chlorogenic acid, cryptochlorogenic acid, scopoletin, isochlorogenic acid B, narigin, isochlorogenic acid A, isochlorogenic acid C and cinnamaldehyde widentified. The linear range of hydrochloride ephedrine, hydrochloride pseudoephedrine, neochlorogenic acid, scopolin, chlorogenic acid, cryptochlorogenic acid, scopoletin, isochlorogenic acid A, isochlorogenic acid C and cinnamaldehyde were 2.475-247.5, 2.600-260.0, 3.820-382.0, 3.900-390.0, 3.060-306.0, 4.760-476.0, 4.540-454.0, 4.900-490.0, 4.540-454.0, 7.590-759.0 μg/mL (r>0.999 0). The limits of quantitation were 0.320 0, 0.350 0, 0.021 4, 0.024 3, 0.036 8, 0.025 7, 0.152 1, 0.042 9, 0.025 1, 0.350 3 μg/mL, respectively;the limits of detection were 0.160 0, 0.180 0, 0.007 9, 0.004 0, 0.001 2, 0.007 3, 0.076 0, 0.001 4, 0.008 1, 0.201 4 μg/mL, respectively. RSDs of precision, stability and reproducibility tests were all lower than 2%. The recoveries were 95.03%-106.85% (RSD were 0.67%-2.68%,n=6).The contents were 0.013 3- 0.214 1 mg/mL. CONCLUSIONS: Established fingerprint is stable, accurate and specific, and can be used for quality control of Fengliaoxing fengshi dieda wine; the content determination method is rapid, accurate and reliable, and can be used for simultaneous determination of 10 components.
KEYWORDS Fengliaoxing fengshi dieda wine; HPLC; Fingerprints; Content determination
冯了性风湿跌打药酒收载于2015年版《中国药典》(一部),该药由丁公藤、麻黄、桂枝等27味中药组成[1]。方中丁公藤祛风湿为主药,配以桂枝、麻黄、羌活、蚕沙、白芷发散风寒、祛风湿、舒筋络,香附、木香、厚朴、枳壳、陈皮、苍术、苦杏仁、猪牙皂行气燥湿化痰,小茴香、当归、川芎、乳香、没药、五灵脂、牡丹皮温经活血祛瘀,白术、山药、补骨脂、黄精、菟丝子、泽泻补肝肾、强腰膝,临床主要用于治疗风寒湿痹(风湿、类风湿)、瘀滞肿痛、跌打损伤诸症,疗效确切[2-3]。目前,2015年版《中国药典》(一部)关于该药的质量标准中未有含量测定和指纹图谱测定项[1],且现有相关文献报道中未有绿原酸类和异绿原酸类等有效成分的检测[4-6]。虽然关红晖等[7]建立了冯了性风湿跌打药酒的高效液相(HPLC)指纹图谱,但存在特征峰数量少、指纹特征不明显等不足。由于冯了性风湿跌打药酒的药效成分较多,因此以单一成分或者单一药材所含的有效成分作为定量测定指标,不能全面控制其内在质量,也无法体现整体质量。
中药指纹图谱是一种综合的、可量化的鉴定手段[8-10],是国际公认的控制中药或天然药材质量的方法[11-12],能够体现中药质量控制的整体性[13-14]。基于此,本研究在前期研究的基础上,建立了冯了性风湿跌打药酒的HPLC指纹图谱,并测定了其中10种有效成分的含量,旨在为其整体质量控制提供参考。
1 材料
1.1 仪器
1260型HPLC仪,包括G7114A紫外检测器、G7129A自动进样器、G7111A四元泵、OpenLAB CDS色谱工作站(美国Agilent公司);KQ-100型超声波清洗机(昆山市超声仪器有限公司);BS210S型、Max210g型万分之一电子分析天平,BT25S、Max21g型十万分之一电子分析天平[赛多利斯科学仪器(北京)有限公司]。
1.2 药品与试剂
冯了性风湿跌打药酒[国药集团冯了性(佛山)药业有限公司,批准文号:国药准字Z44022914,批号:170908、171106、171215、180521、180710、180823、180917、190104、190113、190325,规格:500 mL/瓶;依次编号为S1~S10];盐酸麻黄碱对照品(批号:171241-201809,纯度:≥98%)、盐酸伪麻黄碱对照品(批号:171237-201510,纯度:≥98%)均购自中国食品药品检定研究院;柚皮苷对照品(批号:wkq19012809,纯度:≥98%)、桂皮醛对照品(批号:wkq19031207,纯度:≥98%)均购自四川维克奇生物有限公司;东莨菪苷对照品(批号:16040805)、东莨菪内酯对照品(批号:161208)、绿原酸对照品(批号:1701904)、隐绿原酸对照品(批號:17061401)、新绿原酸对照品(批号:17062003)、异绿原酸A对照品(批号:18090301)、异绿原酸B对照品(批号:17121201)、异绿原酸C对照品(批号:18070401)均购自上海圻明生物科技有限公司(纯度均不低于98%);乙腈、甲醇为色谱纯,其余试剂均为分析纯,水为超纯水。
2 方法与结果
2.1 指纹图谱的建立
2.1.1 色谱条件 色谱柱:Inertsil ODS-3 C18(150 mm ×4.6 mm,5 μm);流动相:乙腈(A)-0.1%磷酸水溶液(B),梯度洗脱(0~15 min,5%A;15~15.01 min,5%A→10%A;15.01~40 min,10%A→15%A;40~70 min,15%A→25%A;70~90 min,25%A→40%A;90~110 min,40%A→70%A;110~120 min,70%A);检测波长:210 nm(10~15 min,盐酸麻黄碱、盐酸伪麻黄碱)、300 nm(70~120 min,桂皮醛)、345 nm(15~70 min,新绿原酸、东莨菪苷、绿原酸、隐绿原酸、东莨菪内酯、异绿原酸A、异绿原酸C、柚皮苷、异绿原酸B);流速:1.0 mL/min;柱温:25 ℃,进样量:10 μL。
2.1.2 混合对照品溶液Ⅰ的制备 精密称取盐酸麻黄碱、盐酸伪麻黄碱、新绿原酸、东莨菪苷、绿原酸、隐绿原酸、东莨菪内酯、异绿原酸B、柚皮苷、异绿原酸A、异绿原酸C、桂皮醛对照品各适量,置于25 mL量瓶中,加甲醇制成上述12种成分质量浓度分别为0.247 5、0.260 0、0.382 0、0.390 0、0.306 0、0.476 0、0.454 0、0.463 0、0.389 5、0.490 0、0.450 0、0.759 0 mg/mL的混合对照品Ⅰ溶液。
2.1.3 供试品溶液的制备 精密量取样品10 mL,置于20 mL量瓶中,加甲醇稀释至刻度,摇匀,经0.45 μm微孔滤膜滤过,取续滤液,即得。
2.1.4 精密度试验 取“2.1.3”项下供试品溶液(编号:S1)适量,按“2.1.1”项下色谱条件连续进样测定6次,以东莨菪内酯峰的保留时间和峰面积为参照,计算各共有峰的相对保留时间和相对峰面积。结果,18个共有峰相对保留时间的RSD为0.29%~1.68%(n=6),相对峰面积的RSD为0.22%~1.27%(n=6),表明本方法精密度良好。
2.1.5 稳定性试验 取“2.1.3”项下供试品溶液(编号:S1)适量,分别于室温下放置0、2、4、8、12、24 h时,按“2.1.1”项下色谱条件进样测定,以东莨菪内酯峰的保留时间和峰面积为参照,计算各共有峰的相对保留时间和相对峰面积。结果,18个共有峰相对保留时间的RSD为0.69%~1.31%(n=6),相对峰面积的RSD为0.51%~1.83%(n=6),表明供试品溶液于室温下放置24 h内稳定性良好。
2.1.6 重复性试验 取样品(编号:S1)约10 mL,共6份,按“2.1.3”项下方法制备供试品溶液,再按“2.1.1”项下色谱条件进样测定,以东莨菪内酯峰的保留时间和峰面积为参照,计算各共有峰的相对保留时间和相对峰面积。结果,18个共有峰相对保留时间的RSD为0.19%~0.81%(n=6),相对峰面积的RSD为0.41%~1.59%(n=6),表明本方法重复性良好。
2.1.7 HPLC指纹图谱的建立 取10批样品,按“2.1.3”项下方法制备供试品溶液,再按 “2.1.1”项下色谱条件进样测定,记录色谱图。采用《中药色谱指纹图谱相似度评价系统(2012版)》,以S4样品(随机选取)色谱图作为对照色谱,设定时间窗为0~120 min,时间宽度为0.3 min,经多点校正后自动匹配,采用平均值法生成HPLC叠加指纹图谱和对照指纹图谱,详见图1、图2。
2.1.8 相似度评价 采用《中药色谱指纹图谱相似度评价系统(2012版)》对10批样品进行相似度分析。结果,10批样品的相似度均大于0.980,提示不同批次样品的相似度较高,生产工艺相对稳定,结果见表1。
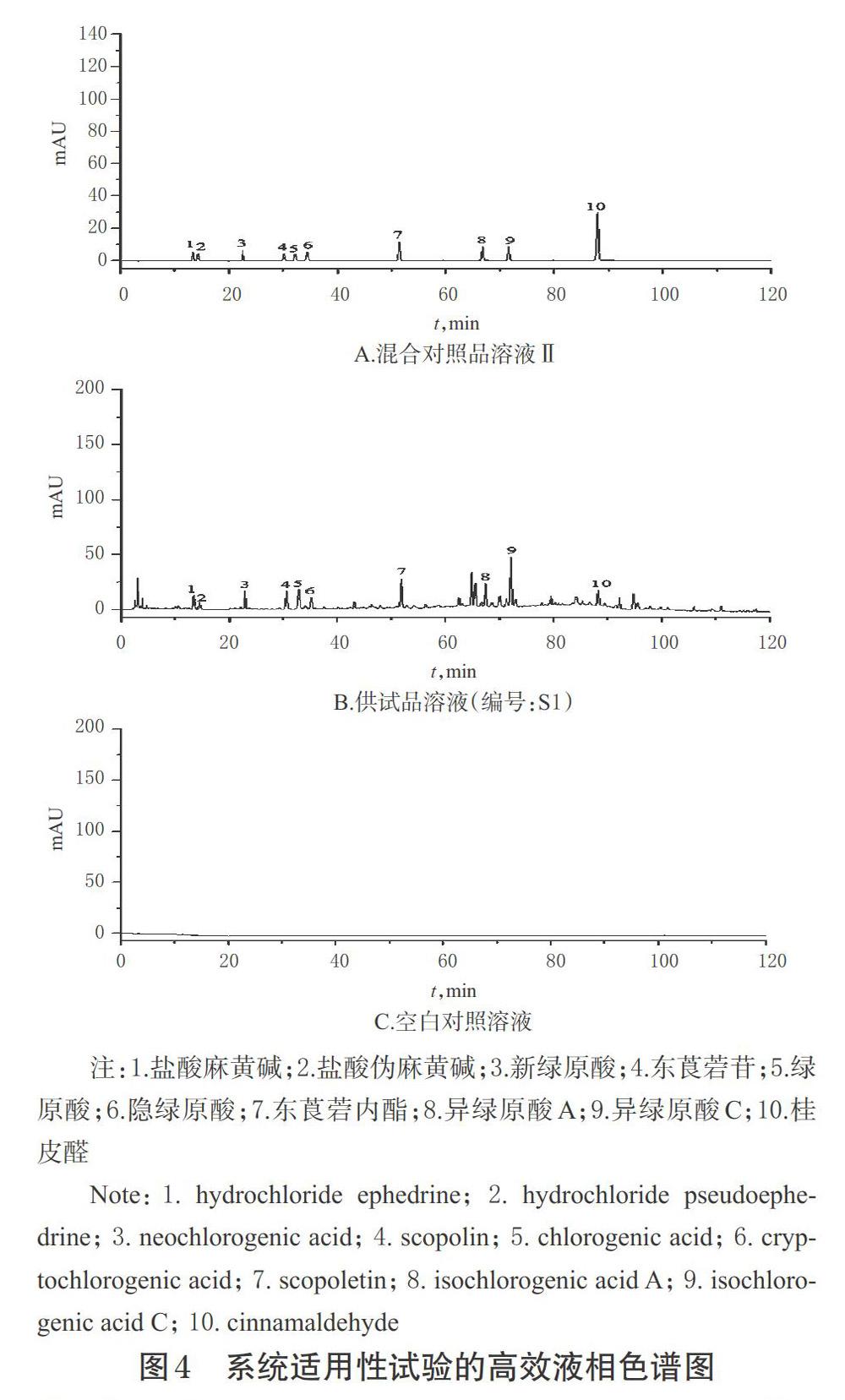
2.1.9 共有峰的指认 10批样品共有18个共有峰,通过与混合对照品溶液Ⅰ(图3)的保留时间比对,指认了12个共有峰,分别为盐酸麻黄碱(1号峰)、盐酸伪麻黄碱(2号峰)、新绿原酸(3号峰)、东莨菪苷(4号峰)、绿原酸(5号峰)、隐绿原酸(6号峰)、东莨菪内酯(8号峰)、异绿原酸B(11号峰)、柚皮苷(12号峰)、异绿原酸A(13号峰)、异绿原酸C(15号峰)、桂皮醛(18号峰)。其中,东莨菪内酯峰(8号峰)峰形好、与相邻色谱峰的分离度好、色谱响应值较高,其对照品价廉易得且保留时间居中,故选择东莨菪内酯峰(8号峰)为参照,计算其他共有峰的相对保留时间和相对峰面积,结果见表2、表3。
2.2 含量测定
2.2.1 色谱条件 同“2.1.1”项。
2.2.2 混合对照品溶液Ⅱ的制备 按“2.1.2”项下方法制备盐酸麻黄碱、盐酸伪麻黄碱、新绿原酸、东莨菪苷、绿原酸、隐绿原酸、东莨菪内酯、异绿原酸A、异绿原酸C、桂皮醛质量浓度分别为0.247 5、0.260 0、0.382 0、0.390 0、0.306 0、0.476 0、0.454 0、0.490 0、0.454 0、0.759 0 mg/mL的混合对照品溶液Ⅱ。
2.2.3 供试品溶液的制备 同“2.1.3”项。
2.2.4 空白对照溶液的制备 精密量取40%乙醇10 mL,置于20 mL量瓶中,加甲醇稀释至刻度,摇匀,经0.45 μm微孔滤膜滤过,取续滤液,即得。
2.2.5 系统适用性试验 取上述混合对照品溶液Ⅱ、供试品溶液(编号:S1)、空白对照溶液各适量,按“2.2.1”项下色谱条件进样测定,记录色谱图。结果,各待測峰与相邻峰间的分离度均大于1.5,理论板数不低于10 000,详见图4。
2.2.6 线性关系考察 精密量取“2.2.2”项下混合对照品溶液Ⅱ0.1、0.5、1、5、10 mL,分别用甲醇定容至10 mL量瓶中,得系列浓度工作溶液,按“2.2.1”项下色谱条件进样测定,记录峰面积。以各待测成分质量浓度(x,μg/mL)为横坐标、峰面积(y)为纵坐标进行线性回归,结果见表4。
2.2.7 定量限与检测限考察 取“2.2.2”项下混合对照品溶液Ⅱ,用甲醇逐级稀释,按 “2.2.1”项下色谱条件进样测定,分别以信噪比10 ∶ 1、3 ∶ 1计算定量限、检测限。结果,盐酸麻黄碱、盐酸伪麻黄碱、新绿原酸、东莨菪苷、绿原酸、隐绿原酸、东莨菪内酯、异绿原酸A、异绿原酸C、桂皮醛的定量限分别为0.320 0、0.350 0、0.021 4、0.024 3、0.036 8、0.025 7、0.152 1、0.042 9、0.025 1、0.350 3 μg/mL,检测限分别为0.160 0、0.180 0、0.007 9、0.004 0、0.001 2、0.007 3、0.076 0、0.001 4、0.008 1、0.201 4 μg/mL。
2.2.8 精密度试验 精密吸取“2.2.2”项下混合对照品溶液Ⅱ适量,按“2.2.1”项下色谱条件连续进样测定6次,记录峰面积。结果,盐酸麻黄碱、盐酸伪麻黄碱、新绿原酸、东莨菪苷、绿原酸、隐绿原酸、东莨菪内酯、异绿原酸A、异绿原酸C、桂皮醛峰面积的RSD分别为0.45%、0.64%、0.91%、0.31%、1.01%、1.14%、0.66%、0.80%、1.11%、0.81%(n=6),表明仪器精密度良好。
2.2.9 稳定性试验 取“2.2.3”项下供试品溶液(编号:S1)适量,分别于室温下放置0、2、4、8、12、24 h时,按“2.2.1”项下色谱条件进样测定,记录峰面积。结果,盐酸麻黄碱、盐酸伪麻黄碱、新绿原酸、东莨菪苷、绿原酸、隐绿原酸、东莨菪内酯、异绿原酸A、异绿原酸C、桂皮醛峰面积的RSD分别为1.76%、0.73%、0.87%、0.49%、0.34%、0.95%、1.23%、1.08%、0.68%、0.71%(n=6),表明供试品溶液在24 h内稳定性良好。
2.2.10 重复性试验 精密称取样品(编号:S1)10 mL,共6份,按“2.2.3”项下方法制备供试品溶液,再按“2.2.1”项下色谱条件进样测定,记录峰面积并按外标法计算样品含量。结果,盐酸麻黄碱、盐酸伪麻黄碱、新绿原酸、东莨菪苷、绿原酸、隐绿原酸、东莨菪内酯、异绿原酸A、异绿原酸C、桂皮醛含量的RSD分别为1.57%、0.42%、0.87%、0.89%、1.45%、1.09%、0.56%、0.63%、0.99%、1.83%(n=6),表明本方法重复性良好。
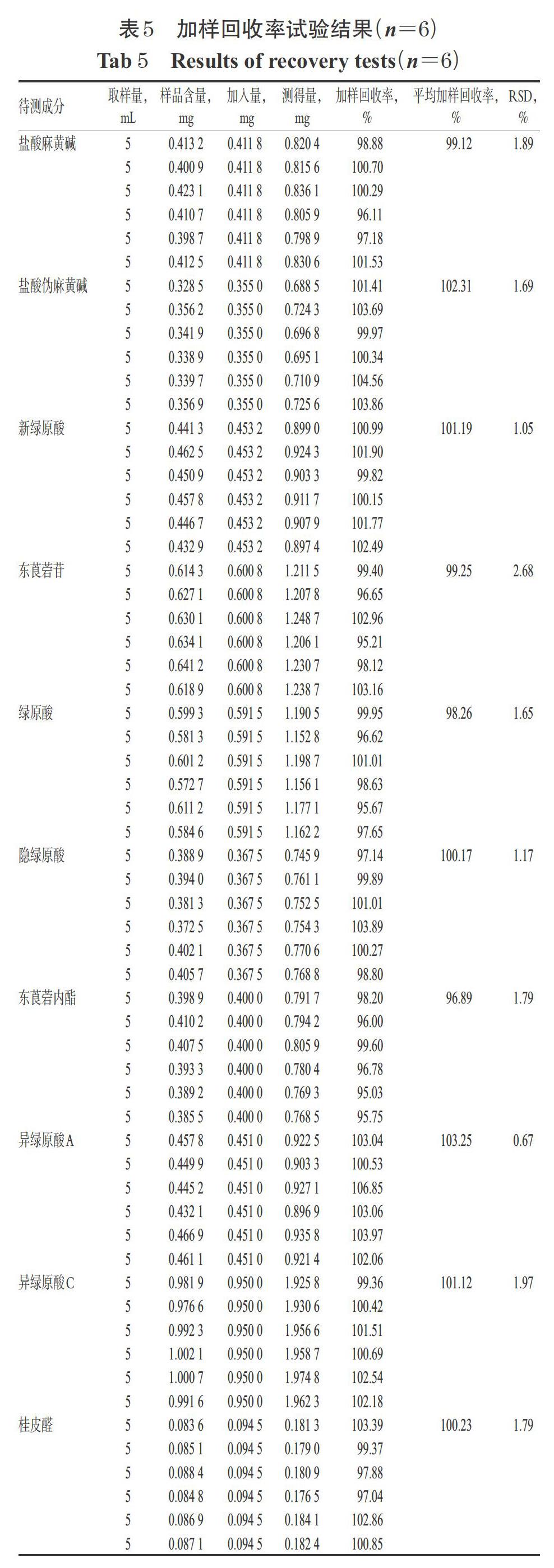
2.2.11 加样回收率试验 精密量取样品(编号:S1)5 mL,置于20 mL量瓶中,共6份,加入一定量的“2.2.1”项下混合对照品溶液Ⅱ,按“2.2.3”项下方法制备供试品溶液,再按“2.2.1”项下色谱条件进样测定,记录峰面积并计算加样回收率。结果,10种待测成分的加样回收率范围为95.03%~106.85%(RSD为0.67%~2.68%,n=6),结果见表5。
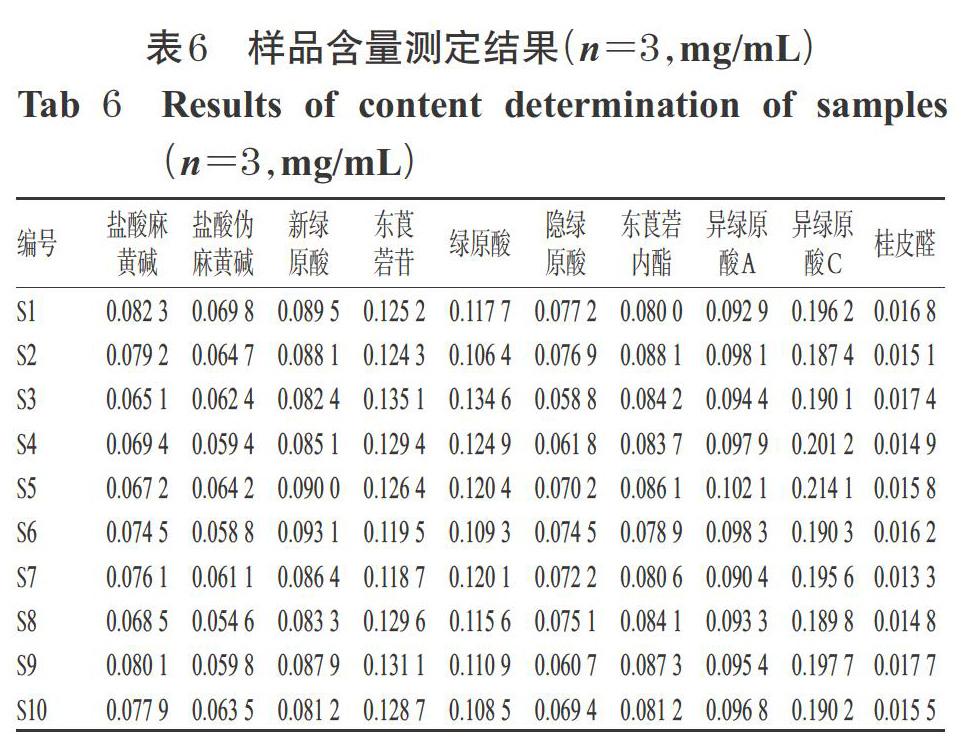
2.2.12 样品含量测定 取10批样品各10 mL,按“2.2.3”项下方法制备供试品溶液,再按“2.2.1”项下色谱条件进样测定,记录峰面积并按外标法计算样品中各成分的含量。各样品平行操作3次,结果见表6。
3 讨论
3.1 检测波长的选择
本研究采用紫外检测器在200~400 nm波长范围内进行扫描,比较各待测成分在不同波长下的色谱图。结果发现,各待测成分的最大吸收波长有显著差异,大部分成分的最大吸收波长在345 nm附近,且分离度较好,响应值较高;虽然在300 nm波长处时色谱峰信息最丰富,但各峰整体响应较345 nm低,吸收不均匀,却可指认桂皮醛色谱峰,故选择300 nm波长检测桂皮醛;盐酸麻黄碱、盐酸伪麻黄碱在210 nm波长处有最大吸收且在其他波长处的紫外吸收急剧下降,为保证共有峰数目多、分离度好、灵敏度高、干扰少,故选择于210 nm波长处检测盐酸麻黄碱、盐酸伪麻黄碱,于300 nm波长处检测桂皮醛,于345 nm波长处检测其余成分。
3.2 参照峰的选择
由于盐酸麻黄碱和盐酸伪麻黄碱的末端吸收干扰大,绿原酸类、异绿原酸类成分及桂皮醛易分解,故选择对照品价廉易得、性质稳定、与其他峰分离度较好且药理作用明确的东莨菪内酯作为参照峰。
3.3 指标性成分的选取
本课题组在前期研究中发现,丁公藤中含有大量的绿原酸类、异绿原酸类和香豆素类成分,尤其是异绿原酸类成分中的异绿原酸B、异绿原酸A、异绿原酸C等含量较高[15],且具有抗炎[16]、抗氧化[17]等药理活性,这些药理活性與丁公藤具有的抗风湿作用有关。有研究证实,麻黄中生物碱类成分具有发汗和解热作用,与桂枝配伍可增强其发汗解肌的功效[18-19]。故选择丁公藤、麻黄和桂枝中的药效成分作为质量控制的指标性成分具有可行性(由于柚皮苷、异绿原酸B的分离度较差,均小于1.5,故未选择其作为定量分析的指标性成分)。
3.4 指纹图谱分析
冯了性风湿跌打药酒的HPLC指纹图谱中共有18个共有峰,本研究指认了其中12个成分,分别为盐酸麻黄碱、盐酸伪麻黄碱、新绿原酸、东莨菪苷、绿原酸、隐绿原酸、东莨菪内酯、异绿原酸B、柚皮苷、异绿原酸A、异绿原酸C及桂皮醛。10批冯了性风湿跌打药酒样品相似度较好,均大于0.980,提示冯了性风湿跌打药酒的原料质量和制备工艺较稳定,不同批次样品中指标性成分的一致性较好。
3.5 含量测定结果分析
本研究结果显示,盐酸麻黄碱等10种有效成分为不同批次冯了性风湿跌打药酒的共有成分,但各批样品间含量存在较大差异,特别是绿原酸和异绿原酸C的含量相差约0.01 mg/mL。因此,建议将这10种有效成分含量纳入样品的质量标准,可提高其整体质量。
综上所述,本研究所建指纹图谱稳定、准确、专属性强,可用于冯了性风湿跌打药酒的质量控制;所建含量测定方法简便快速、准确可靠,可用于同时测定该药中10种有效成分的含量。
参考文献
[ 1 ] 国家药典委员会.中华人民共和国药典:一部[S]. 2015年版.北京:中国医药科技出版社,2015:808-809.
[ 2 ] 谭珍,曹颖珍,王永健.冯了性风湿跌打药酒真伪品及掺假品的鉴别[J].中国中医药信息杂志,2006,13(9):56- 57.
[ 3 ] 姚血明,马武开,唐芳,等.中医外治法在痹证中的应用[J].风湿病与关节炎,2014,3(5):73-75.
[ 4 ] 查道成,冯冬兰,李林晓,等. HPLC测定冯了性风湿跌打药酒中盐酸麻黄碱的含量[J].中国实验方剂学杂志,2013,19(9):129-131.
[ 5 ] 杨媛媛,陈志永,廖立平,等.冯了性风湿跌打药酒中东莨菪素、东莨菪苷和绿原酸的测定[J]. 中成药,2014,36(3):551-553.
[ 6 ] 李海燕,谢仕伟,岑志芳. HPLC法测定冯了性风湿跌打药酒中东茛菪内酯的含量[J].佛山科学技术学院学报,2009,27(3):75-77.
[ 7 ] 关红晖,霍嘉茵,林淑明,等.冯了性风湿跌打药酒HPLC指纹图谱研究[J].广东药科大学学报,2017,33(1):65- 67.
[ 8 ] XIE RF,ZHAO QH,LI ZC,et al. Comparison on HPLC fingerprints between fraxini cortex and its eye drop[J]. Chin Herb Med,2013,5(4):301-306.
[ 9 ] 刁嘉茵,徐灿,王淑美,等.中药指纹图谱与药效相关性研究进展[J].药学研究,2018,37(3):165-168.
[10] 陈振华,刘苏珍,唐芳,等.浅谈中药质量标准现状与几种质量评价方法[J].时珍国医国药,2016,27(3):694-696.
[11] 徐妍,杨华蕊,杨永寿,等.中药指纹图谱研究现状及展望[J].世界最新医学信息文摘,2018,18(76):91-94.
[12] GAO SM,LIU JS,WANG M,et al. Quantitative and HPLC fingerprint analysis combined with chemometrics for quality evaluation of Codonopsis radix processed with different methods[J]. Chin Herb Med,2019,11(2):160- 168.
[13] 张婷,郑夺,王文彤,等.指纹图谱结合一测多评模式在参芎养心颗粒质量评价中的应用研究[J].中草药,2015,46(13):1920-1925.
[14] 吴雯,吴锐枫,康江丽.参苓健体粉指纹图谱和含量测定方法的研究[J].中国药师,2019,22(5):852-856.
[15] CHEN ZY,LIAO LP,YANG YY,et al. Different fingerprinting strategies to differentiate Porana sinensis and plants of erycibe by high-performance liquid chromatography with diode array detection,ultra high performance liquid chromatography with tandem quadrupole mass spectrometry,and chem[J]. J Sep Sci,2015,38(2):231-238.
[16] 那襲雪,张文涛,谈远锋,等.绿原酸及其异构体药理作用及不良反应研究进展[J].辽宁中医药大学学报,2018,20(3):140-144.
[17] CHEN ZY,TAO HX,LIAO LP,et al. Quick identification of xanthine oxidase inhibitor and antioxidant from Erycibe obtusifolia by a drug discovery platform composed of multiple mass spectrometric platforms and thin-layer chromatography bioautography[J]. J Sep Sci,2014,37(16):2253-2259.
[18] 王艳宏,王秋红,夏永刚,等.麻黄化学拆分组分的性味药理学评价:化学拆分组分的制备及其解热作用的研究[J].中医药信息,2011,28(5):7-10.
[19] 王艳宏,王秋红,夏永刚,等.麻黄化学拆分组分的性味药理学评价:麻黄化学拆分组分“辛温”发汗、利水作用的实验研究[J].中国中医药科技,2011,18(6):489-491.
(收稿日期:2019-09-24 修回日期:2020-03-16)
(编辑:陈 宏)
