误诊为系统性红斑狼疮的皮肤型Rosai-Dorfman-Destombes病一例
2020-04-30么艳茹王吉波温大蔚
么艳茹 王吉波 张 婷 温大蔚
1青岛大学附属医院风湿免疫科,山东青岛,266003;2山东大学齐鲁医院(青岛)风湿科,山东青岛,266035
皮肤型Rosai-Dorfman-Destombes病是一种良性组织细胞增生性疾病,临床上非常罕见。我科诊治1例曾误诊为系统性红斑狼疮而最终经皮肤活检确诊为皮肤型Rosai-Dorfman-Destombes病的患者,现报道如下。
临床资料患者,女,60岁。因“面部对称性红斑1年,加重伴丘疹、斑块形成半年”来诊。患者于1年前无明显诱因出现双侧面部对称性红斑,大小约4.0 cm×5.0 cm,类似于系统性红斑狼疮的面部蝶形红斑(图1a),无疼痛、瘙痒,偶有双膝关节疼痛。曾于外院查ANA阳性,滴度1∶320,CRP及ESR升高,血常规及肝肾功均无异常,膝关节X线检查提示无关节骨破坏,拟诊为“系统性红斑狼疮”,予泼尼松30 mg/日,硫酸羟氯喹0.2 g/日口服,治疗3个月症状无缓解,且双侧红斑面积逐渐扩大,遂自行停药。半年前,于红斑基础上出现米粒大红棕色丘疹,丘疹逐渐增多并出现结节,局部融合成斑块,同时背部出现约3.0 cm×3.0 cm的单个类似皮损,患者自行涂抹药膏(具体不详)后,皮损表面出现渗出、结痂和脱屑,过程中无皮肤溃疡、疼痛和瘙痒,现为进一步明确诊治来诊。患者有系统性红斑狼疮家族史(患者妹妹的女儿患有“系统性红斑狼疮”),无其他特殊病史。
体格检查:全身浅表淋巴结未触及肿大,肝脾未触及肿大。皮肤情况:双侧面颊、下颌及鼻背部皮肤大片红斑,其上及周围可见散在或成簇分布的米粒大红棕色硬性丘疹、结节(图1b),局部融合成斑块(图1c),触之硬,有浸润,无压痛;部分丘疹结节表面呈脓疱样改变,红斑表面可见轻微渗出。后背部皮肤可见一约3.0 cm×3.0 cm红斑,其上散在红棕色丘疹(图1d),无压痛。
辅助检查:血尿常规、肝肾功、血糖及血脂等均正常;C-反应蛋白25.36 mg/L(正常值:<5 mg/L);血沉42.00 mm/h(正常值:<20 mm/h);抗核抗体阳性,滴度1∶320;抗dsDNA抗体、抗ENA抗体谱均为阴性;EBV及HIV等病毒学相关检测均为阴性;血清IgG4为0.2 g/L(正常值:0.03~2.01 g/L);CT扫描未发现淋巴结或其他器官病变。考虑该患者皮疹并非SLE的典型皮疹表现,且临床表现与实验室检查结果并不符合SLE的分类诊断标准,诊断不明确,遂取面部皮损行组织病理学检查:皮肤真皮深层及皮下组织弥漫性淋巴浆细胞浸润,其中混合大的组织细胞,形成深浅不一的结节状结构,细胞有伸入运动(emperipolesis)(图2);免疫组织化学特征性表现为S-100、CD68阳性和CD1a阴性(图3)。
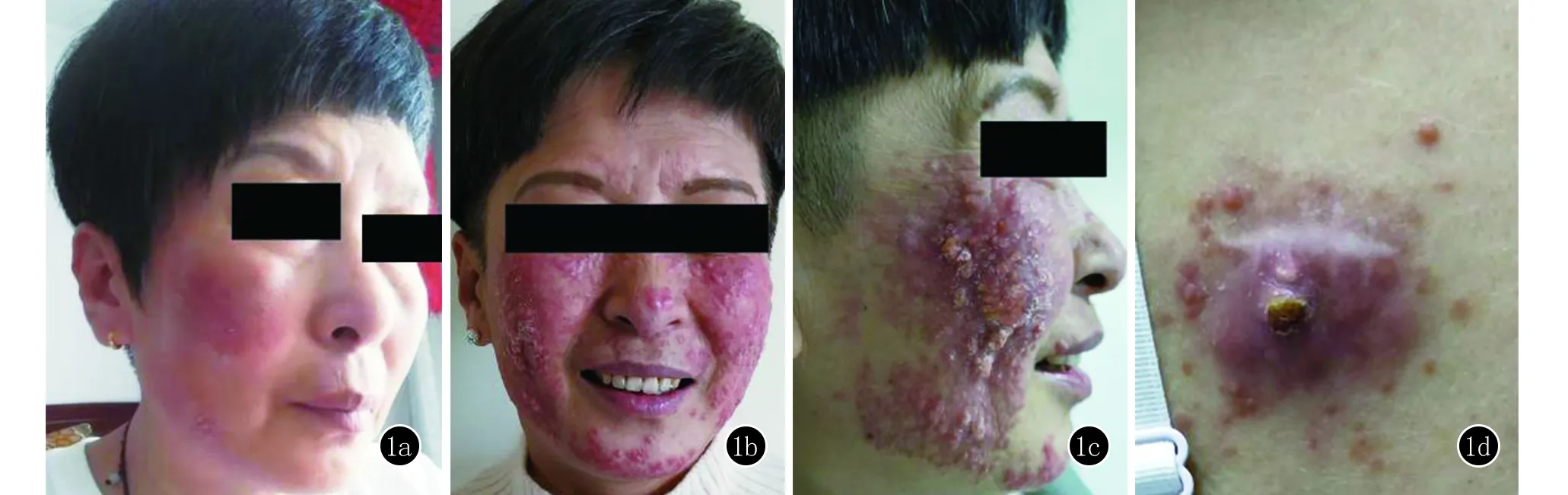
图11a:右侧面颊部约4.0 cm×5.0 cm红斑;1b:双侧面颊、下颌及鼻背部皮肤大片红斑,其上及周围可见散在或成簇分布的米粒大红棕色硬性丘疹、结节;1c:局部丘疹融合成斑块, 部分丘疹结节表面呈脓疱样改变,红斑表面可见轻微渗出;1d:后背部皮肤可见一约3.0 cm×3.0 cm红斑,其上散在红棕色丘疹
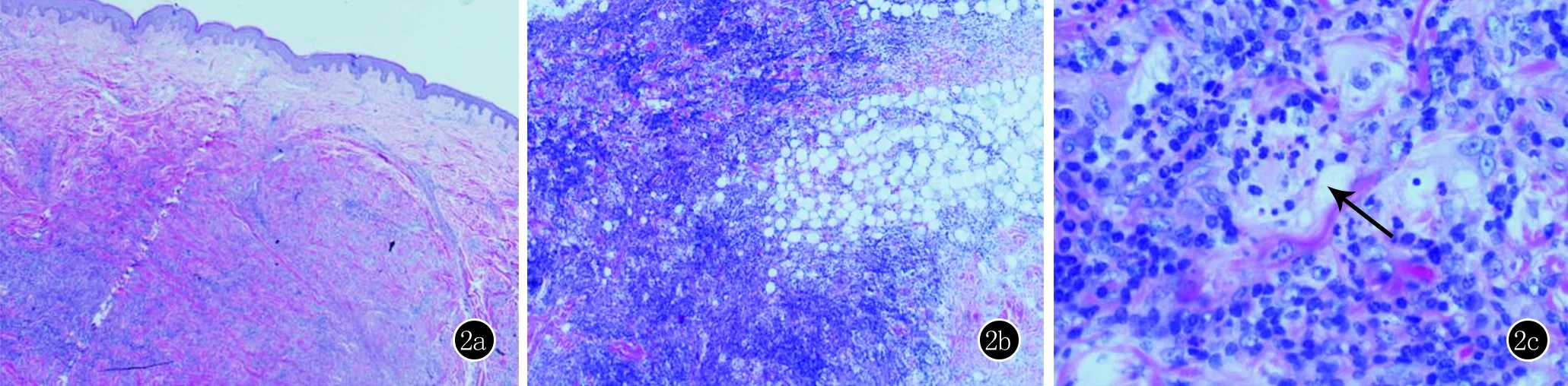
图22a:真皮深层及皮下组织弥漫性小淋巴细胞、浆细胞等炎性细胞浸润(HE,×40);2b:炎性细胞中可见成片增生的组织细胞,胞体巨大,两者形成深染区与淡染区交错的不规则结节状结构(HE,×100);2c:高倍镜下可见组织细胞吞噬完整的淋巴细胞、中性粒细胞等炎性细胞,即“伸入运动”(箭头所示)(HE,×400)
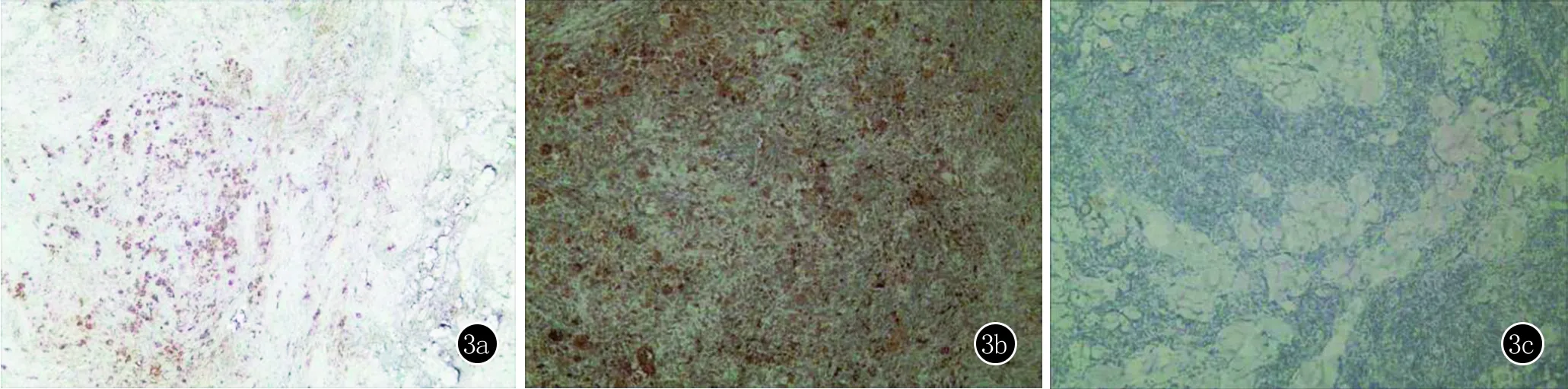
图3 3a:S-100蛋白阳性(免疫组化,×100);3b:CD68阳性(免疫组化,×100);3c:CD1a阴性(免疫组化,×100)
诊断:皮肤型Rosai-Dorfman-Destombes病(CRDD)。治疗:泼尼松25 mg/日,甲氨蝶呤10 mg/周,沙利度胺25 mg/日口服,泼尼松逐渐减量。治疗期间无明显药物不良反应,治疗2个月后皮损面积较前减小、颜色变淡、硬度下降,未见新发皮损,目前随访中。
讨论Rosai-Dorfman-Destombes病(RDD)是一种罕见的非朗格汉斯组织细胞增生性疾病,该病最早于1965年由法国病理学家Destombes报道[1],1969年Rosai和Dorfman对此病进行总结,并命名为窦组织细胞增生伴巨大淋巴结病(sinus histiocytosis with massive lymphadenopathy,SHML)[2]。根据临床特点,该病被分为经典(淋巴结)RDD和结外RDD两种类型[3]。结外RDD可与淋巴结病变同时发生,也可作为唯一的首发部位,结外所有的系统及器官均可受累,但以皮肤受累最常见,而单纯发生于皮肤无系统损伤者非常罕见,约占所有RDD的3%,称为皮肤型Rosai-Dorfman-Destombes病(cutaneous Rosai-Dorfman-Destombes disease,CRDD)。
目前,CRDD的病因及发病机制尚不明确。研究发现CRDD可能与病毒感染(EBV、HIV及HPV-6等)、IgG4相关性疾病及SLC29A3基因突变等有关。CRDD多见于亚裔女性,平均患病年龄45.2岁[4]。CRDD皮损通常表现为生长缓慢的、无痛性、非瘙痒性丘疹、结节或斑块,其颜色可为黄色、红色或棕色,全身各处皮肤均可发生,以四肢、颜面最为常见。临床上CRDD需要与寻常痤疮、皮肤结核、皮肤淋巴瘤等疾病鉴别。但面部CRDD常见受累部位为面颊部,且病变多呈双侧分布[5],故面部皮肤受累时,亦应与系统性红斑狼疮(systemic lupus erythematosus,SLE)进行鉴别诊断。SLE患者皮疹常为颧部呈蝶形分布的红斑、盘状红斑、指掌部和甲周红斑、指端缺血等,其中以鼻梁和双颧颊部呈蝶形分布的红斑最具特征性;而CRDD患者皮损如上所述,以丘疹、结节和斑块为主。据统计,10%的RDD与免疫性疾病(如SLE等)共存,称为免疫相关性RDD[3]。RDD合并SLE的患者,除淋巴结增大及皮疹等RDD的表现外,尚可有光过敏、口腔溃疡、浆膜炎、关节炎、蛋白尿、溶血性贫血甚至狼疮脑病等SLE独有的临床表现。Alqanatish等[6]曾报道1例经组织病理学确诊的RDD患者,3年后该患者出现心慌、气促、心包积液,心包穿刺后于心包积液中发现狼疮细胞,进一步查血dsDNA、ANA阳性、补体低下、三系下降、Coombs试验阳性,最终确诊为RDD合并SLE。因此,确诊RDD的患者也有必要进行血清ANA、RF、抗dsDNA抗体、抗ENA抗体谱及免疫球蛋白等免疫指标的检测[3],以筛查相关的自身免疫性疾病。对于本例患者,SLE的家族史、起初的面部对称性红斑、偶发的膝关节疼痛及血清ANA阳性等因素导致该患者于外院就诊时被误诊为SLE,而于我院进一步查抗dsDNA抗体、抗ENA抗体谱及免疫球蛋白等指标均为阴性,排除了SLE的诊断。
通常情况下,CRDD的诊断主要依靠组织病理并结合免疫组化染色,其显著的病理特点为增生的组织细胞吞噬淋巴细胞、浆细胞及中性粒细胞等炎性细胞,即伸入运动(emperipolesis);组织细胞免疫组化:S-100、CD68阳性,CD1a阴性。在病理表现上,CRDD需要与朗格汉斯组织细胞增生症等其他组织细胞增生性疾病相鉴别。本例患者仅出现面部及背部皮损,无淋巴结肿大,系统检查未见异常,组织病理可见伸入运动,免疫组化示组织细胞S-100、CD68阳性,CD1a阴性,符合CRDD的诊断。
CRDD是良性疾病,20%~50%的患者可自发缓解,故部分病人确诊后可随访观察[3]。而对于症状明显或病变影响外观的患者,可选择手术、糖皮质激素、化学治疗、免疫调节疗法,靶向治疗及放射治疗等治疗方案[3]。有研究统计表明[4,5],手术切除局部病变是效果最好的治疗手段,59%的病人可完全缓解,而对于本例患者,其面部皮疹严重影响外观但面积大无法手术切除,遂予口服泼尼松联合甲氨蝶呤、沙利度胺方案。目前来看初步治疗效果尚可,但皮疹能否完全消除仍需临床随访观察。国内亦有其他应用糖皮质激素联合免疫抑制剂成功治疗CRDD的病例报道[7,8]。所以,临床上在选择治疗方法时应当根据病情而定,必要时可联合应用上述多种治疗方案。
CRDD临床少见且皮疹缺乏特异性,容易误诊及漏诊,本文报道了1例起初被误诊为SLE而最终经皮肤活检确诊的CRDD患者。本病例的诊疗过程提示:疑诊SLE的患者面部红斑不典型时需进一步行组织病理学检查与CRDD相鉴别;同时,对于确诊CRDD的患者,亦需行进一步的免疫学检查以筛查可能并存的SLE等免疫性疾病。
