锂电池正极材料ε-VOPO4的第一性原理计算
2020-04-27李娜,苏蓉,赵辉
李 娜,苏 蓉,赵 辉
(天津师范大学物理与材料科学学院,天津300387)
随着电子设备及电动汽车的发展,人们对锂离子电池性能的要求越来越高.作为一种可替代的正极材料,磷酸钒(VOPO4)已被评估为锂离子电池和钠离子电池的潜在候选材料[1].VOPO4具有优异的脱嵌锂离子的性能,可以形成一系列同质多晶化合物[2].这些化合物均由VO6八面体和PO4四面体相互连接构成,每种同质多晶体中八面体和四面体基团间的连接方式不同,目前报道的结构包括层状结构的 αⅠ、αⅡ、ω、δ 和γ 以及三维结构的 β 和 ε 共 7 种[2].唐安平等[3]报道了VOPO4各种晶型的初始可逆容量和放电电压,指出初始容量随晶型不同而差别很大,放电电压均不低于3.7 V,尤其是ε-VOPO4的放电电压高达3.95 V.Chen等[4]以V2O5和 H3PO4为原料合成了 ε-VOPO4,其首次充放电比容量高达227.9 mAh/g.Nicholas 等[5]利用X 线光电子能谱(XPS)和硬X 线光电子能谱(HAXPES)对电化学放电引起的ε-VOPO4电极界面相变进行化学分辨和深度分辨,证明第2 次锂反应在第1 次锂完全掺入前开始.Lin 等[6]首次证明固相合成的ε-LiVOPO4具有超过20 个周期的稳定循化, 并结合DFT 计算发现ε-LiVOPO4可能是一种低导电性的准一维离子扩散体.本研究利用基于密度泛函理论(DFT)的第一性原理,对ε-VOPO4锂电池正极材料在单个锂原子嵌入过程中引起的晶格结构和电子性能变化进行研究.
1 计算模型与方法
ε-VOPO4具有由VO6八面体与PO4四面体通过角共享连接形成的一维链组成的三维隧道结构[7].图1为ε-VOPO4的球棍模型和多面体晶体结构图.由图1(a)可以直观地看出ε-VOPO4晶体中各个原子的空间分布情况,由图1(b)可以看出完整的VO6八面体及PO4四面体结构.在 ε-VOPO4单胞中,V(红色)原子的数量和P(蓝色)原子的数量为4,O(黄色)原子的数量为20.ε-VOPO4晶体中,相邻V 原子与O 原子的距离约为0.190 0 nm.相邻P 原子与O 原子间的距离约为0.150 0 nm.以P 原子为顶点的相邻的2 个P—O 键的夹角为60°,即由P—O 键构成的四面体为正四面体.
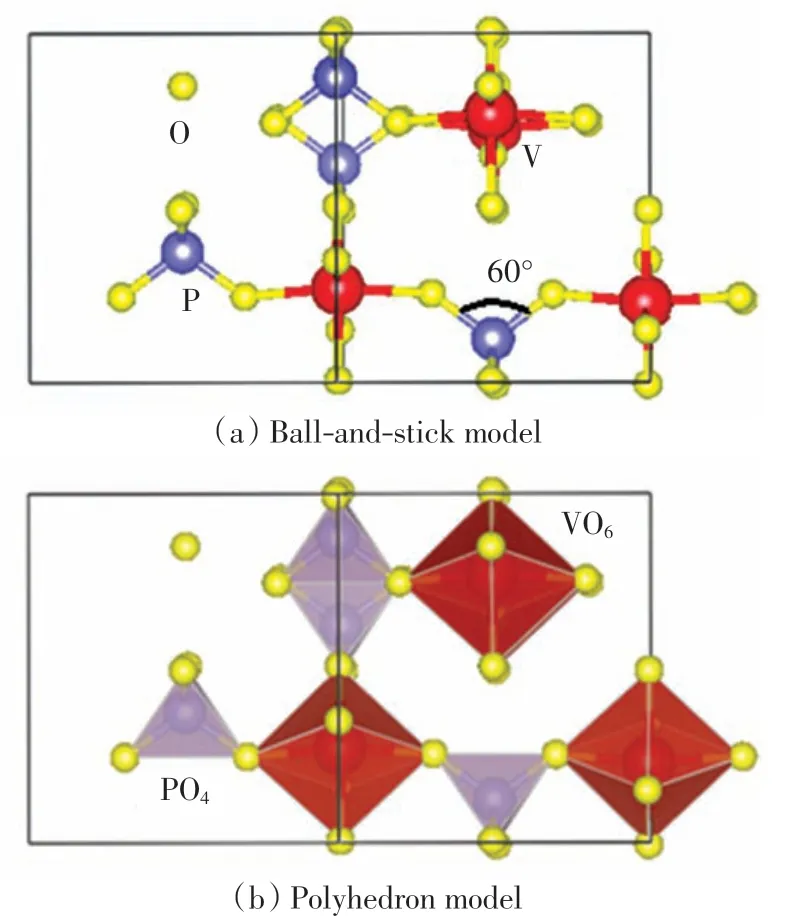
图1 ε-VOPO4 的晶体结构Fig.1 Crystal structure of ε-VOPO4
本研究利用基于密度泛函的第一性原理计算方法对作为锂离子电池正极材料的ε-VOPO4的性能参数进行理论研究.首先根据ε-VOPO4的结构特点对其嵌锂行为进行第一性原理研究.通过ε-VOPO4的晶体结构分析体系中锂的嵌入位置,并利用结构优化与能量计算方法, 得到体系中锂的稳定嵌入位置.然后对连续嵌入锂的ε-VOPO4体系进行第一性原理能量计算,通过体系的能量计算结果得到体系的形成能以及能带带隙随嵌锂数量的变化关系.采用Material studio 8.0 软件包中的CASTEP 程序进行结构的优化及结构形成能和电子性质的计算.CASTEP 是基于密度泛函理论(DFT)平面赝势方法的量子力学程序.在计算中,选用广义梯度近似(GGA)下的PBE 泛函来描述电子间的交换相关能, 根据Mokhorst-pack 方法对K 点进行采样.平面波截止能取500 eV, 不可约布里渊区的K 点取3×3×3, 体系中各原子核内层电子对外层电子的库仑吸引势计算采用超软赝势(ultrasoft pseudo potential).在进行各项计算前均采用BFGS 方法对构建的模型进行几何优化,以获得最稳定的结构.进行自洽计算时,原子总收敛能取2×10-5eV/atom,平均原子力低于0.5 eV/nm,最大原子位移容差小于2×10-2nm,选择 1s22s1、3d34s2、2s22p4和 3s23p3作为 Li、V、O 和 P的价电子构型.由于GGA 方法在描述过渡金属V 的3d态电子时误差较大,本研究采取GGA+U 方法,经过测试U 取值为3.5 eV.
为了测试赝势平面波计算的可靠性, 对ε-VOPO4晶胞使用3×3×3 的K 点网格和500 eV 的平面波截断能进行收敛测试.计算所得晶格参数如表1 所示.由表1 可以看出,本研究所得计算值与文献[8]中实验值的误差均小于1%,说明所选参数是合适的.
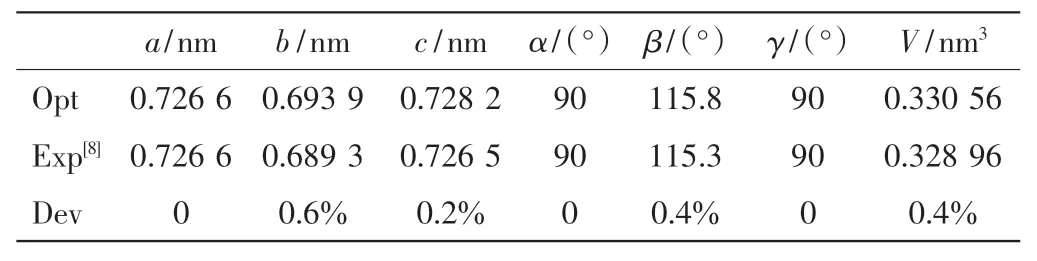
表1 ε-VOPO4 实验与优化的晶格参数Tab.1 Experimental and optimized lattice parameters of ε-VOPO4
2 计算结果与讨论
2.1 嵌插位置
为了准确了解锂的插层位置,计算每个可能的嵌插位置后, 找到最可能的嵌插点.图2 为嵌入锂离子后 ε-VOPO4的晶体结构,其中图2(a)为在 ε-VOPO4单胞中嵌入一个Li 原子(绿色)的 ε-L(iVOPO4)4晶体的球棍模型结构,图2(b)为嵌入2 个锂原子(绿色)的ε-Li(2VOPO4)4晶体的球棍模型结构.
经过第一性原理结构优化计算,由图2 可以清晰地看出,Li 的最佳嵌插点位于V-O 八面体与P-O 四面体之间的间隙, 与氧形成一个四面体.同样通过角共享与V-O 八面体与P-O 四面体连接,这与Chen 等[8]的实验结果一致.
2.2 形成能
物质的形成能可以表征物质形成的难易程度及其稳定性.理论上,只有物质的形成能小于0,反应才能进行[9].因此,本研究计算了ε-VOPO4单胞结构连续嵌锂的形成能,分析了ε-VOPO4单胞对锂的吸附性能.连续嵌锂的形成能可以表示为

式(1)中:x 为锂在 ε-VOPO4中的原子比;Ef为体系的形成能;Ex为嵌入的物质的量浓度为x 的ε-Lix(VOPO4)4的形成能;E0为未嵌入锂ε-VOPO4的结构的形成能;E1为单个锂原子的形成能[10].进行第一性原理计算后得到单胞中嵌入1 个锂原子和2 个锂原子优化后的结构的形成能分别为-0.54 eV 和-6.94 eV,说明嵌锂后结构是稳定存在的,且随着Li 的嵌入化合物的形成能降低.
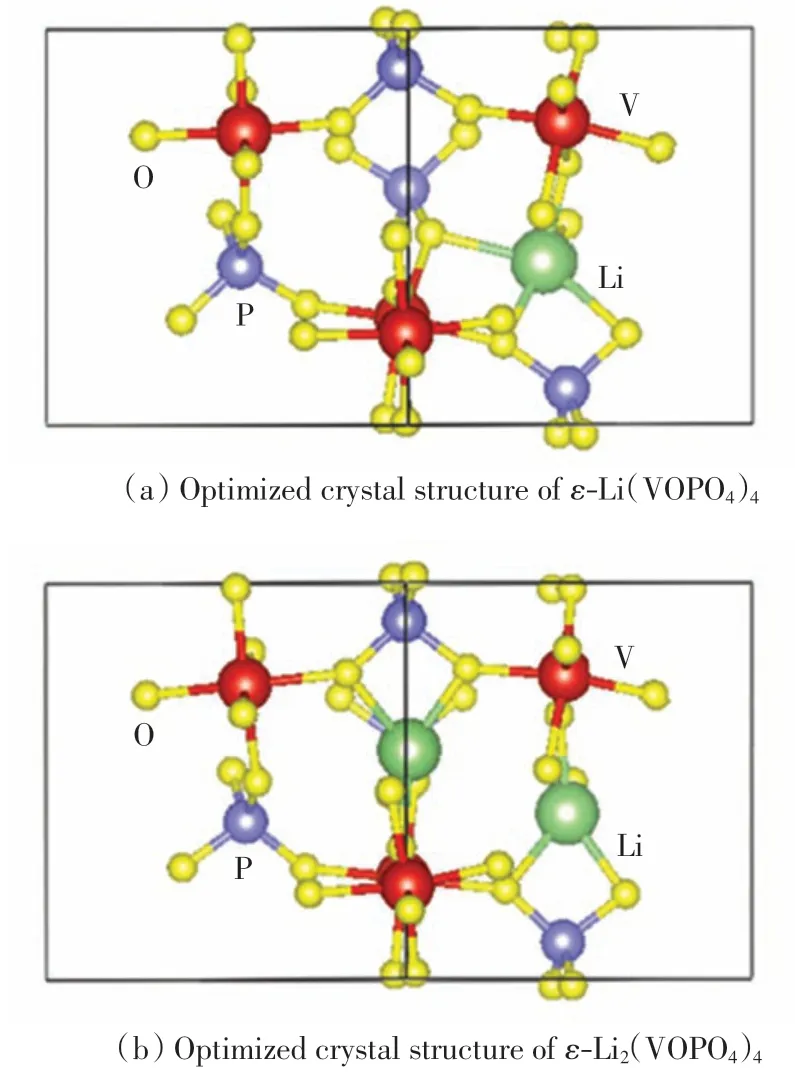
图2 ε-Lix(VOPO4)4(x=1 和2)的晶体结构Fig.2 Crystal structure of ε-Lix(VOPO4)4(x=1 and 2)
2.3 电子能带结构
固体材料根据禁带宽度可以分为导体、半导体和绝缘体3 类.在禁带宽度的基础上, 根据固体材料的能带结构可以确定其导电性[11].因此,能带结构可以提供有关锂化磷酸钒电子结构的重要信息,ε-VOPO4及嵌入Li 后的能带结构如图3 所示.由图3 可知, 作为一种半导体材料,ε-VOPO4的带隙为2.009 eV,而ε-Li(VOPO4)4和ε-Li2(VOPO4)4的带隙值分别为0.895 eV和 1.048 eV.与 ε-VOPO4相比,锂化后 ε-VOPO4的带隙值降低,说明电子很容易从价带激发到导带,而电子导电率的增加改善了电化学性能[12].此外,锂化的ε-VOPO4的能带结构向能量低的方向移动,表明锂嵌入ε-VOPO4的稳定性良好.综上所述, 锂的嵌入降低了ε-VOPO4的能带间隙,增强了ε-VOPO4的导电性.
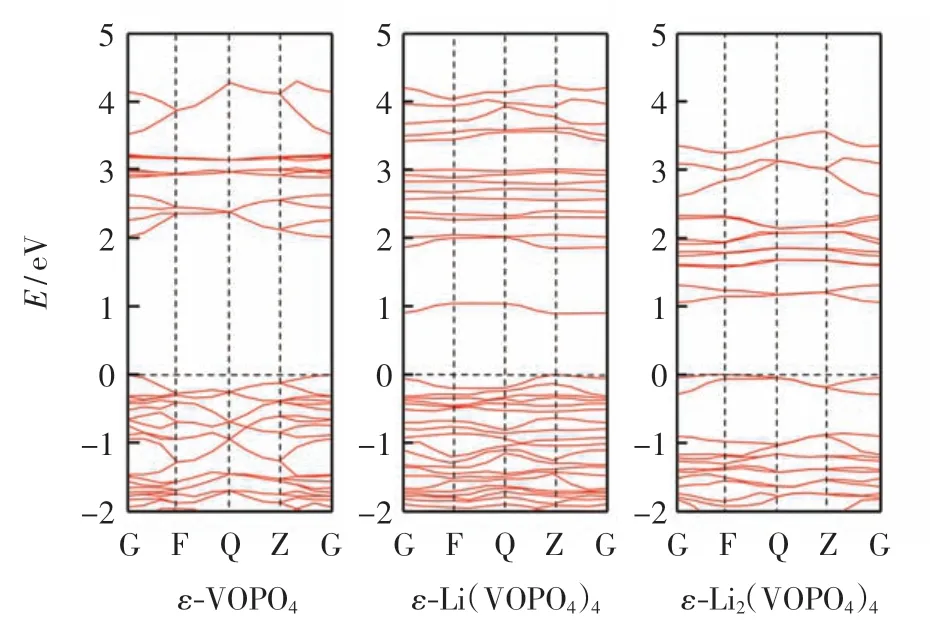
图3 ε-Lix(VOPO4)4(x=0、1 和2)的能带结构Fig.3 Electronic band structures of ε-Lix(VOPO4)4(x=0,1 and 2)
2.4 电子态密度分析
ε-VOPO4、ε-Li(VOPO4)4和ε-Li2(VOPO4)4的晶胞分态密度和总态密度图如图4 所示.由图4(a)可以看出,价带分布在-65~0 eV,导带分布在0~10 eV,ε-VOPO4的导带主要由O2s和V3d组成.在价带-40 ~-37 eV 处出现了很高的尖峰, 这是因为作为过渡态金属,V 离子的态密度相对比较局域,而在-20 eV 处出现的尖峰是 O2s、P3s和 V3d共同贡献的.由图4(b)和图4(c)可知,锂的嵌入导致整个结构的导带向低能量方向移动,但并没有与其他原子形成强的杂交重叠,反映了锂原子与其相邻原子间的弱相互作用,这有利于锂原子在充放电过程中的移动.由于锂原子的嵌入,-65 eV周围的态密度分布发生变化, 峰值由20 eV-1降低到12 eV-1.导带峰值由45 eV-1降低到38 eV-1,价带的峰值由25 eV-1减小到13 eV-1.这些变化说明锂原子的嵌入使得ε-VOPO4化合物的导电性增强[13].此外,与Li1s带相比,O2p带和V3d带对锂化后ε-VOPO4费米能级附近的DOS 贡献更大.图4 中没有观察到Li1s与O2p态之间的强杂交, 这说明Li 原子与O 原子间为弱相互作用,而锂原子与其相邻原子间的弱相互作用有利于锂原子的脱层和插层[13].
2.5 电荷布居分析
为了阐明不同原子之间的相互作用,表2 给出了ε-Lix(VOPO4)4化合物的电荷布局分布和键的布居分布.在ε-Lix(VOPO4)4中,5 个不等价氧原子(O1、O2和O3、O4、O5)分别与金属原子的相互作用不同,因此本文取平均值代表不同O 原子与金属原子间的相互作用[14].由表2 可知,锂原子的净电荷(1.03 e、1.06 e)与其形式电荷(+1 e)非常接近,说明Li 离子与O 离子间为纯离子键,意味着高电离度有利于Li 离子在ε-VOPO4中的循环脱嵌.V 离子净电荷(1.44 e、1.33 e)与其形式电荷(+4 e)的偏差表明ε-Lix(VOPO4)4中V 离子与O 离子间分别形成了强共价键.P 离子净电荷(2.34 e、2.29 e)与形式电荷(+4 e)的偏差表明ε-Lix(VOPO4)4中的P离子与O 离子和V 离子与O 离子一样形成了强共价键.通过计算键的布居分布可以定量评估键的强度,正键和负键分别代表成键态和反键态, 键能越大,共价度越高,而其值接近0 表示2 个原子间没有明显的相互作用[15].Li—O 键的键能(-0.1 e、-0.02 e)接近 0表明ε-Lix(VOPO4)4中的Li 与O 间没有明显的相互作用.V—O 键的键能在嵌入1 个Li 原子和2 个Li 原子后分别减小了0.007 e 和0.002 e,这说明在Li 的嵌入过程中V—O 键没有发生变化.而P—O 键的键能在嵌入1 个Li 原子和2 个Li 原子后分别减小了0.003 e和0.004 e,这说明在Li 的嵌入过程中P—O 键也没有发生变化.综上所述,在锂离子的脱嵌过程中,ε-VOPO电极材料的结构具有较高的稳定性.
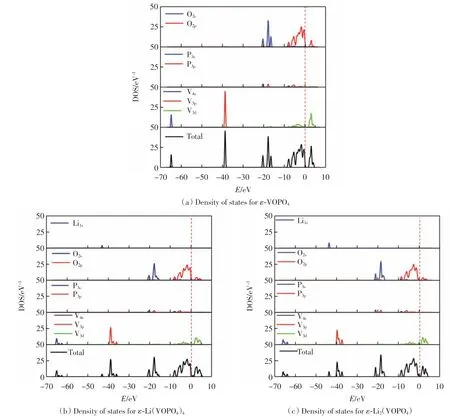
图4 ε-Lix(VOPO4)(x=0、1 和2)的密态度结构Fig.4 Density of states(DOS)4 of ε-Lix(VOPO4)4(x=0,1 and 2)
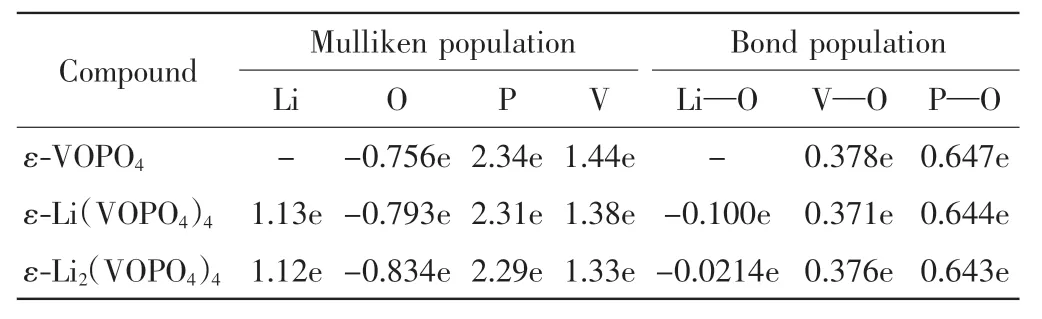
表2 ε-Lix(VOPO4)4(x=0、1 和2)的电荷布居和键布居分布Tab.2 Mulliken population and bond population for ε-Lix(VOPO4)4(x=0,1 and 2)
3 结论
本文采用第一性原理计算方法,研究了作为锂离子电池正极材料的ε-VOPO4在锂嵌入过程中引起的电化学变化,计算结果表明:
(1)由计算所得形成能可知, 嵌锂后的结构是可以稳定存在的,且随着Li 原子的嵌入,结构的形成能更低.最佳嵌插点位于V-O 八面体与P-O 四面体之间的间隙.
(2)Li 原子嵌入ε-VOPO4中会使材料的能带带隙减小,费米能级向能量低的方向移动有益于提高电极材料的导电率.
(3)布居分析表明ε-Lix(VOPO4)4(x=1,2)中Li离子和O 离子间没有明显的相互作用,Li 与相邻原子间的弱相互作用有利于Li 的脱层和插层.随着Li 原子的嵌入,V—O 键键能和P—O 键键能并没有发生大的变化,说明电极材料具有较高的稳定性.
