脐带间充质干细胞制剂的质量管理及有效性和安全性
2020-04-27王佃亮
王佃亮
国内外利用脐带间充质干细胞制剂进行的动物模型试验、临床研究和临床移植治疗表明,不同的脐带间充质干细胞制剂疗效差别很大[1-5]。究其原因,可能与疾病种类、治疗时机、使用剂量、移植途径、个体差异等因素有关,但主要还是由干细胞制剂的质量差异导致的。造成脐带间充质干细胞制剂质量差异的因素很多,主要包括细胞来源的组织材料、分离制备方法及过程、检测鉴定方法及过程、传代方法及过程、扩增方法及过程、制剂配制添加成分及配制方法和过程、冷藏或冷冻保存方法及过程、复苏方法及过程、操作人员技能差异等。干细胞制剂的质量与干细胞治疗的有效性和安全性有关,是干细胞临床应用的关键。
1 脐带间充质干细胞制剂的质量管理
临床级的干细胞制剂以人体疾病治疗为目的,其生产和管理应遵循《药品生产质量管理规范》(good manufacturing practice,GMP)、《人体细胞治疗研究和制剂质量控制技术指导原则》(国家食品药品监督管理局2003年3月颁布)、《干细胞制剂质量控制及临床前研究指导原则(试行)》(国家卫生计生委、食品药品监督管理总局2015年7月颁布)、《干细胞制剂制备质量管理自律规范》(中国医药生物技术协会2016年10月发布)、《干细胞通用要求》(中国细胞生物学学会干细胞生物学分会2017年11月发布)等国家法规和行业标准[6-7]。在脐带间充质干细胞制剂生产过程中,需要严格遵守这些法规和标准,提高质量。
1.1 制剂制备过程及所用材料的质量管理 预先制定新生儿脐带采集及脐带间充质干细胞分离纯化、原代培养、传代扩增、细胞鉴定、细胞系建立、冷冻保存、干细胞注射液配制和保存的标准操作及质量管理程序,建立质量标准(表1),并在符合GMP要求基础上严格执行。
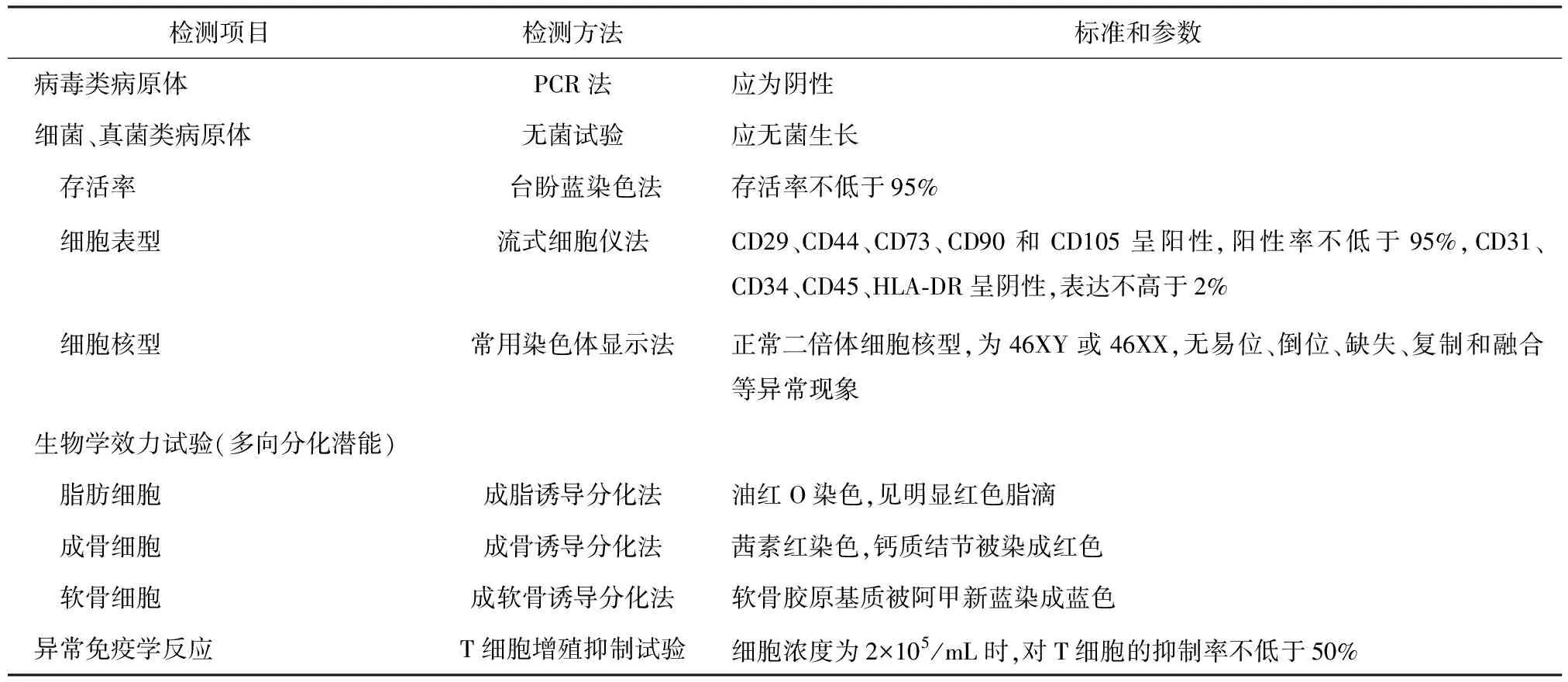
表1 脐带间充质干细胞的质量标准
新生儿脐带样本的采集场所应达到Ⅱ级一般洁净手术室要求,保证无菌采集环境。采集前,选择健康足月产妇,保证:①获得产妇本人、法定代表人或监护人同意,签署知情同意书;②经检验产妇乙型肝炎病毒(hepatitis B virus,HBV)、丙型肝炎病毒(hepatitis C virus,HCV)、艾滋病病毒(human immunodeficiency virus,HIV)、EB病毒(Epstein-Barr virus,EBV)、巨细胞病毒(cytomegalovirus,CMV)、人类嗜T淋巴细胞病毒(human T cell lymphotropic virus,HTLV)、梅毒螺旋体、支原体、霉菌等病原体均为阴性;③产妇本人及家族成员无遗传病、传染病、精神病或其他重大疾病现病史和既往病史。采集时,待胎儿娩出,立刻用蘸有75%酒精的纱布擦拭脐带,截取至少15 cm无针孔的脐带放入保存液中,置于无菌采集袋里,标识清楚,脐带两端用手术线结扎。
在无菌室操作台上,从新生儿脐带组织剥取华通氏胶,以组织块培养法或酶消化法分离新生儿间充质干细胞,贴壁法做原代培养。培养液中若加有牛血清、猪胰酶,除要求供应商提供企业合法生产资质、产品合格证及说明书等信息外,还要检测牛、猪来源的危险致病病毒。传代培养要有明确的细胞鉴别特征,细胞纯度和活性达到临床要求,并且无内外源微生物污染。经传代和扩增培养后达到一定数量的细胞系,可以进行冷冻保存。冻存细胞需加入合适的冷冻保护液,在程序降温条件下进行冷冻。冻存好的细胞移至液氮罐中(-196~-135 ℃)长期储存。建立主细胞库和工作细胞库,减少不同批次脐带间充质干细胞在操作过程中的变异性[8]。
干细胞培养液添加物、干细胞注射液配制所需的溶媒和添加物都要有合法的来源,生产企业有相应资质,能够提供产品合格证和说明书等信息。干细胞制备及制剂配制的相关重要信息(如:干细胞供者的年龄、民族、血型、现病史、既往病史等;干细胞制剂的生产日期、批号、有效期、入库时间、出库时间等)应录入计算机系统进行管理,核心数据信息要进行备份,以防感染计算机病毒或遭黑客袭击丢失信息。
1.2 制剂的质量检测 临床移植治疗用的脐带间充质干细胞制剂,需要经过细胞检测、制剂检测、放行检测以及质量复核等程序。
1.2.1 细胞检测 包括但不限于:①细胞鉴别:通过细胞形态、遗传学、代谢酶亚型谱分析、表面标志物、特定基因表达产物等检测,对不同供体的脐带间充质干细胞进行综合的细胞鉴别。在倒置显微镜下观察,体外微组织块培养法培养的原代人脐带间充质干细胞在第6~8天开始有少量细胞从组织块中爬出,沿组织周围向外呈放射状延伸生长,吹打去除组织块后呈旋涡状生长,细胞呈梭形,与皮肤成纤维细胞类似,形态相对均一。原代人脐带间充质干细胞长满瓶底并达到90%融合时间为18~20 d。传代培养的脐带间充质干细胞生长速度比原代快,24 h内可见少量贴壁的圆型细胞,逐渐呈克隆式生长,到3 d左右时,大多数呈梭形,部分呈多边或多角形,长满瓶底后基本呈梭形,旋涡状生长。根据其生长形态和特征,结合组织来源,可初步鉴别脐带间充质干细胞。在通过表面标志物等分析,可明确鉴定。正常二倍体细胞核型,为46 XY或46 XX;CD29、CD44、CD73、CD105、CD90阳性,阳性率大于95%;HLA-DR、CD34、 CD31、CD45阴性,表达小于2%等。②存活率和生长活性:采用多种细胞生物学活性检测方法,如活细胞计数、细胞倍增时间、细胞周期、克隆形成率、端粒酶活性等判断细胞活性和生长情况。存活率不低于95%;体外扩增人脐带间充质干细胞潜伏期为1~2 d,对数生长期为3~7 d,第8天以后长满瓶底转入平台期,生长停滞,传代培养增殖倍数为5~6倍;人脐带间充质干细胞85%以上处于G0/G1期,G2/M期占5%以下,S期细胞占10%以下。③纯度、均一性及最低装量:通过肉眼观察、检测细胞表面标志物和遗传多态性及特定生物学活性等,对制剂进行细胞纯度或均一性的检测;用最低装量检查法检查最低装量。标准和参数为白色均匀悬浊液,无明显沉淀,无异物。每剂的最低装量应为(20±2)mL。④无菌试验和细菌、真菌、支原体、梅毒螺旋体检测:依据2015版《中华人民共和国药典》中生物制品无菌试验和支原体检测规程,对细菌、真菌、支原体污染进行检测。标准和参数为:应无菌生长;细菌、支原体、真菌、梅毒螺旋体检测为阴性。⑤细胞内外源致病因子检测:应结合体内和体外方法,根据每一细胞制剂的特性进行人源及动物源性特定致病因子,利用PCR技术、酶联免疫法等进行检测。如使用过牛血清,须进行牛源特定病毒的检测;如使用胰酶等猪源材料,应至少检测猪源细小病毒。另外,还应检测逆转录病毒(如HIV)。标准和参数为:HBV、HCV、HIV、EBV、CMV、HTLV、牛海绵状脑病病毒、猪细小病毒等检测应为阴性。⑥内毒素检测:依据2015版《中华人民共和国药典》中内毒素检测规程中的凝胶法,对内毒素进行检测。内毒素检测标准为≤50 EU。⑦异常免疫学反应检测:检测异体来源干细胞对人总淋巴细胞增殖和对不同淋巴细胞亚群增殖能力的影响,或对相关细胞因子分泌的影响,分析可能引起的异常免疫反应。判定标准为间充质干细胞浓度为2×105/mL时,对T淋巴细胞增殖抑制率不低于50%。⑧致瘤性和促瘤性检验:对于异体来源的干细胞制剂或经体外复杂操作的自体干细胞制剂,须通过免疫缺陷动物体内致瘤试验,检验细胞的致瘤性。判定标准为无致瘤性。⑨生物学效力试验:通过检测干细胞分化潜能、诱导分化细胞的结构和生理功能、对免疫细胞的调节能力、分泌特定细胞因子、表达特定基因和蛋白等功能,判断干细胞制剂与治疗相关的生物学有效性。对间充质干细胞,无论何种来源,应进行体外多种类型细胞(如成脂肪细胞、成软骨细胞、成骨细胞等)分化能力的检测,以判断其细胞分化的多能性。除此以外,作为特定生物学效应试验,应进行与其治疗适应证相关的生物学效应检验。判定标准为应具有相应生物学功能。⑩细胞培养基及其他添加成分残余量的检测:对脐带间充质干细胞制备过程中残余的、影响脐带间充质干细胞制剂质量和安全性的成分,如牛血清白蛋白、抗生素、细胞因子等进行检测。注射剂中牛血清残留量检测使用牛血清白蛋白酶联免疫法或间接凝集法,判定标准为牛血清残留量不得大于50 ng/mL;细胞注射剂抗生素残留量检测采取培养法,标准为阴性;人表皮生长因子采用ELISA的方法检测,应不大于100 pg/mL。
1.2.2 制剂检测 脐带间充质干细胞制剂(注射液)是脐带间充质干细胞与0.9%的生理盐水或复方电解质注射液(基础溶媒)、L-谷氨酰胺或腺苷(能源物质)、N-乙酰半胱氨酸或亚硒酸钠(抗氧化剂)、胰岛素(活性生长因子)、肝素钙(抗凝剂)、甘油或二甲亚砜(渗透性细胞内冷冻保护剂)、人血白蛋白或葡聚糖(非渗透性细胞外冷冻保护剂)等成分配制而成。它的检测是在干细胞检测的基础上,进一步检测制剂中脐带间充质干细胞的形态、存活率、生长活性等,检测制剂的纯度和均一性、微生物病原体、内毒素、异常免疫反应、致瘤性、促瘤性、生物学效力等。脐带间充质干细胞注射剂的包装必须采取符合2015版《中华人民共和国药典》的包装要求,包装材料必须无菌、无色透明、不吸附细胞,不影响细胞活性;制备的细胞注射剂成品外观肉眼观察应为白色均匀混悬液,无明显的沉淀物、无异物;每剂的最低装量应为(20±2)mL。
1.2.3 放行检测 针对脐带间充质干细胞制剂的特性,制定放行检测项目和标准。放行检测项目应能在相对短的时间内,反映脐带间充质干细胞制剂的质量和安全信息。
1.2.4 质量复核 由专业细胞检验机构或实验室进行脐带间充质干细胞制剂的质量复核检验,并出具正规的检验报告。
1.3 制剂的稳定性 脐带间充质干细胞制剂的稳定性是指制剂在储存(液氮冻存和细胞植入前的临时存放)和运输过程中的物理、化学和生物学性质的改变。检测项目包括细胞形态、活性、存活率、密度、纯度、无菌性等。稳定性检测的关键是制剂中脐带间充质干细胞的数量和活性的改变,直接影响到脐带间充质干细胞治疗的效果。
根据脐带间充质干细胞制剂稳定性检测结果,确定制剂的保存液成份与配方、保存和运输条件、有效期,同时确定与有效期相适应的运输容器和工具,以及合格的细胞冻存设施和条件。
2 脐带间充质干细胞制剂的有效性
脐带间充质干细胞制剂的有效性可利用细胞模型和动物模型在临床前研究阶段进行评估[9]。①细胞模型通过检测脐带间充质干细胞的分化潜能、诱导分化细胞的结构和生理功能、对免疫细胞的调节能力、分泌特定细胞因子、表达特定基因和蛋白等功能,判断脐带间充质干细胞制剂与治疗相关的生物学有效性。对脐带间充质干细胞进行体外多种类型细胞(成脂肪细胞、成软骨细胞、成骨细胞等)分化能力的检测,以判断细胞分化的多能性。作为特定生物学效应试验,应进行与治疗适应证相关的生物学效应检验。随着研究的进展,针对临床治疗的适应证,会不断研究更新生物学效应检测方法。如研究介导临床治疗效应的关键基因或蛋白的表达,并以此为基础提出与预期的生物学效应相关的替代性生物标志物。②动物模型用于观察植入的脐带间充质干细胞或其分化产物改变模型中疾病的病理进程;研究脐带间充质干细胞的归巢能力和免疫调节功能;通过分析脐带间充质干细胞植入后特定细胞因子和(或)特定基因表达情况,提出替代性生物学效应标志物。
国内外众多临床研究和治疗病例表明,脐带间充质干细胞可用于自身免疫性疾病、退行性疾病、神经性疾病、衰老性疾病、遗传缺陷、组织器官损伤、放射病、炎症等病症的治疗[10-12],包括儿童移植物抗宿主病、克罗恩病、创伤、缺血性损伤、造血功能障碍、骨及软骨损伤、神经损伤、肌营养不良、糖尿病及其并发症、骨关节炎、类风湿性关节炎、系统性红斑狼疮、肝硬化、肝纤维化、酒精性肝病、肝功能衰竭、肾功能衰竭、急性心肌梗死、脑梗死、肿瘤、视网膜黄斑变性、帕金森病、老年痴呆症、肌萎缩侧索硬化症、系统性硬化症、原发性干燥综合征、皮肌炎等难治性疾病。此外,脐带间充质干细胞是比较理想的组织工程种子细胞,可用于体外或体内再生组织器官,进行临床治疗。
3 脐带间充质干细胞制剂的安全性
脐带间充质干细胞制剂的安全性包括生物学安全、病原生物安全、伦理学安全等[9]。脐带间充质干细胞来源于产妇产后的脐带组织,通常情况下是作为医疗废物丢弃的,从脐带组织中分离提取干细胞不存在伦理争议,可以用于临床治疗,在伦理学上是安全的,这一点与某些胚胎干细胞不同[13-15]。
脐带间充质干细胞制剂的生物学安全主要是指毒性反应、异常免疫学反应、非预期分化、致瘤性和促瘤性等。①毒性反应:通过合适的动物实验模型,观察脐带间充质干细胞制剂各种可能的毒性反应,譬如,细胞植入时和植入后的局部和整体的毒性反应。②异常免疫学反应:通过体外和动物试验评价其异常免疫反应,包括对不同免疫细胞亚型及相关细胞因子的影响。③非预期分化:包括非靶细胞分化或非靶部位分化,可利用特定的检测技术,在体内动物试验中研究、评估和监控脐带间充质干细胞非预期分化的可能性。④致瘤性和促瘤性:目前,普遍认为间充质干细胞“不致瘤”或具有“弱致瘤性”,但不排除对已存在肿瘤的“促瘤性”作用,应设计相应的试验方法,以判断制剂的“促瘤性”。对高代次或经过体外复杂处理和修饰的脐带间充质干细胞制剂,应当进行动物致瘤性和促瘤性评估。可选择合适的动物实验模型,使用合适数量的干细胞、合理的植入途径和足够长的观察期,以有效评价制剂的致瘤性和促瘤性。
脐带间充质干细胞制剂的病原生物安全是指携带和传播细菌、病毒、支原体、衣原体、真菌、寄生虫的风险和内毒素残留等。一些常见的病原体,如HBV、HCV、HIV、EBV、CMV、HTLV、梅毒螺旋体、支原体、霉菌、内毒素等,已经建立了实验室检测方法和相应的质量标准,基本可以满足临床要求。随着新的病原体被发现,将来会增加新的检测项目,确保脐带间充质干细胞制剂不含任何内部的和外源的病原体。
通过制定符合临床治疗要求的脐带间充质干细胞制剂生产、储存、运输的质量标准,建立标准化的质量检测方法和流程,进行严格的质量管理,并不断完善这些质量标准、质量检测方法和质量管理流程,脐带间充质干细胞制剂可以达到临床治疗要求。
迄今,国内外大量临床研究和治疗病例表明,脐带间充质干细胞治疗是安全的,除极少数患者轻度腰部酸痛、发热、头昏、头痛外,无其他任何严重不良反应。但是,目前仍有一些患者不宜进行脐带间充质干细胞治疗。这些患者是:晚期恶性肿瘤尤其是脑肿瘤患者;休克或全身衰竭生命体征不正常及不配合检查者;合并心、肺、肝、肾等重要脏器功能障碍者;全身感染或局部严重感染抗感染康复前;凝血功能障碍者;血清学检查(如艾滋病、乙肝、梅毒等)阳性;高度过敏体质或有严重过敏史者;非神经系统疾患或尚未明确诊断者;其他不适于脐带间充质干细胞移植的人群。
