X射线荧光光谱法测定萤石中氟化钙
2020-04-20马景治
马景治
(湖北省地质实验测试中心, 湖北 武汉 430034)
萤石也称氟石,在工业生产中常作为熔剂,主要用于转炉或高炉炼钢的造渣,同时它还是制造氢氟酸以及其它氟化物的基础原料,此外,萤石也是玻璃制造工业上制造隔音和光学玻璃的原料之一.萤石的主要成分为氟化钙(CaF2)、二氧化硅(SiO2)和碳酸盐,其中氟化钙的含量决定了萤石的质量等级[1].萤石中氟化钙的测定主要采用EDTA滴定法[2-3],样品经过乙酸分离碳酸钙,采用不同试剂提取残渣中的氟化钙,加入三乙醇胺掩蔽铁、铝等干扰元素,氢氧化钾调节酸度,EDTA溶液滴定实现测定.该方法准确度高,但对于CaF2含量较低的样品易受镁等离子的干扰导致滴定终点不易准确判断.近年来,采用电感耦合等离子体发射光谱法(ICP-OES)测定氟化钙的方法已有报道[4].该方法用氯化铝(AlCl3)提取氟化钙,采用基体匹配的方法进行测定,与传统方法相比操作简单、快速,然而原子发射光谱法谱线干扰严重,分析结果容易受到不同基体的影响.
X射线荧光光谱法(XRF)测定土壤、岩石、沉积物等样品中的主次量组分较为快速、简单,已被广泛的应用于地质、环境等各个领域[5].采用该法测定萤石中的钙[6-9],以熔融制样,XRF测定萤石中的主、次组分,可以快速、准确分析总钙的含量.但此法测定时,样品中的碳酸钙会计入氟化钙的量,分析结果中未除去其他形式的钙,测定结果准确性较差.因此,采用XRF法测定萤石中氟化钙时,样品需要进行酸处理以除去碳酸钙、铜和铅等元素的干扰.但样品经过酸处理后,不同样品的剩余量不同,造成熔剂与样品的比例不确定,仍然不能准确测定氟化钙的含量.
本研究对样品前处理、XRF分析中熔片条件和仪器工作参数等进行了优化,建立了熔融制样、XRF测定萤石中氟化钙的方法.该方法为测定氟化钙含量提供了新的解决思路.
1 试验部分
1.1 仪器与工作条件
XRF-1800顺序扫描式波长色散X射线荧光光谱仪(日本岛津公司),4.0 kW端窗铑靶X射线光管,75 μm铍窗;真空光路,扫描角度7~148 °.萤石中钙及主要元素的分析条件如表1所列.

表1 XRF最佳测量条件
Optima 8000电感藕合等离子体发射光谱仪(珀金埃尔默仪器有限公司);BS124S分析天平(赛多利斯科学仪器有限公司,北京,感重量0.1 mg);Analymate-V4D高频感应熔样机(北京静远世纪科技有限责任公司),同时熔融两个样品;铂金坩埚(95% Pt+5% Au).
1.2 主要试剂
乙酸、硝酸铵、溴化锂、碘化铵(均为分析纯);二氧化硅(光谱纯);四硼酸锂+偏硼酸锂+氟化锂混合熔剂(分析纯,质量比45∶10∶5),650 ℃灼烧2 h,冷却后置于干燥器中备用;萤石成分分析国家一级标准物质(编号GBW07250~GBW07253),岩石成分分析国家标准物质(编号GBW07108和GBW07120),锌矿石成分分析国家标准物质(编号GBW07237),萤石成分分析外检样品(编号外检-1~外检-12).
1.3 样品分析
准确称取0.300 0±0.000 2 g样品,于105 ℃烘箱中干燥2 h,置于100 mL烧杯中,加入8 mL 10%的乙酸溶液,盖上表面皿,于低温电炉上水浴溶解30 min(期间加水防止蒸干).取下后用水吹洗表面皿及杯壁,冷却至室温.用慢速定量滤纸过滤,用去离子反复水洗后将滤纸和沉淀一起置于30 mL干净的瓷坩埚中.置于马弗炉中微开炉门,从低温升起于700 ℃灼烧30 min,取出后放置在干燥器中.冷却至室温后转移到称量皿称量灼烧物质量,以二氧化硅补加到0.300 0 g,置于原瓷坩埚中,依次称取6.000 0±0.000 4 g四硼酸锂+偏硼酸锂+氟化锂混合熔剂(质量比45∶10∶5)和0.300 0±0.010 0 g硝酸铵于坩埚中,搅匀,转移到铂金坩埚中,加入0.5 mL 0.5 g/mL的溴化锂溶液,于高频熔样机上650 ℃预氧化2 min , 1 050 ℃静置熔融4 min,摇动熔融3 min,冷却后将样片倒出,上机测定.
1.4 标准系列
选用4个萤石国家一级标准物质(编号GBW07250~GBW07253)、两个岩石国家标准物质(编号GBW07108和GBW07120)以及人工配制的校准样品来建立校准曲线.由于萤石的标准物质较少,为了提高基体组成的代表性,选用岩石标准样品GBW07108和GBW07120,分别与上述4个萤石标准物质按照1∶1混合称量,制得8个新的标准样品参与校正,从而构成一套具有一定梯度又有足够的含量范围的标准系列.参与校正的样品无需酸处理,计算校准样品中钙的含量换算为氟化钙的量以建立曲线,熔样方法按照1.3节进行.
2 结果与讨论
2.1 样品制备方法
除了标准曲线样品无需酸处理外,其他样品均需酸处理以避免碳酸钙的影响.本文通过试验对乙酸用量、样品与熔剂稀释比例、脱模剂及熔融温度等的条件进行优化,选择适合该系统分析的最佳样品制备方法.
2.1.1 乙酸用量的选择
萤石中除了含有氟化钙外,常常伴生组分中含有碳酸钙、铜、铅、锌等成分[10].碳酸钙的存在会直接影响总钙的测定,从而使氟化钙测定结果偏高,而铜、铅、锌等金属元素的存在则对铂金坩埚不利.因此,需要对样品进行酸处理,考虑到氟化钙的溶解度,本文选取乙酸进行处理.为了尽可能除去伴存的金属元素,本文对乙酸的用量进行选择.以锌矿石GBW07237作为考察样品,按照选定的处理方法进行溶解,过滤后定容采用ICP-OES测定溶液中Cu、Pb、Zn元素的含量,计算方法的溶出率,结果如图1所示.由图1可见,随着乙酸用量的增加,3个元素的溶出率均会上升,当用量超过8 mL后溶出率变化不大.因此,本方法选择8 mL 10%的乙酸溶解样品.
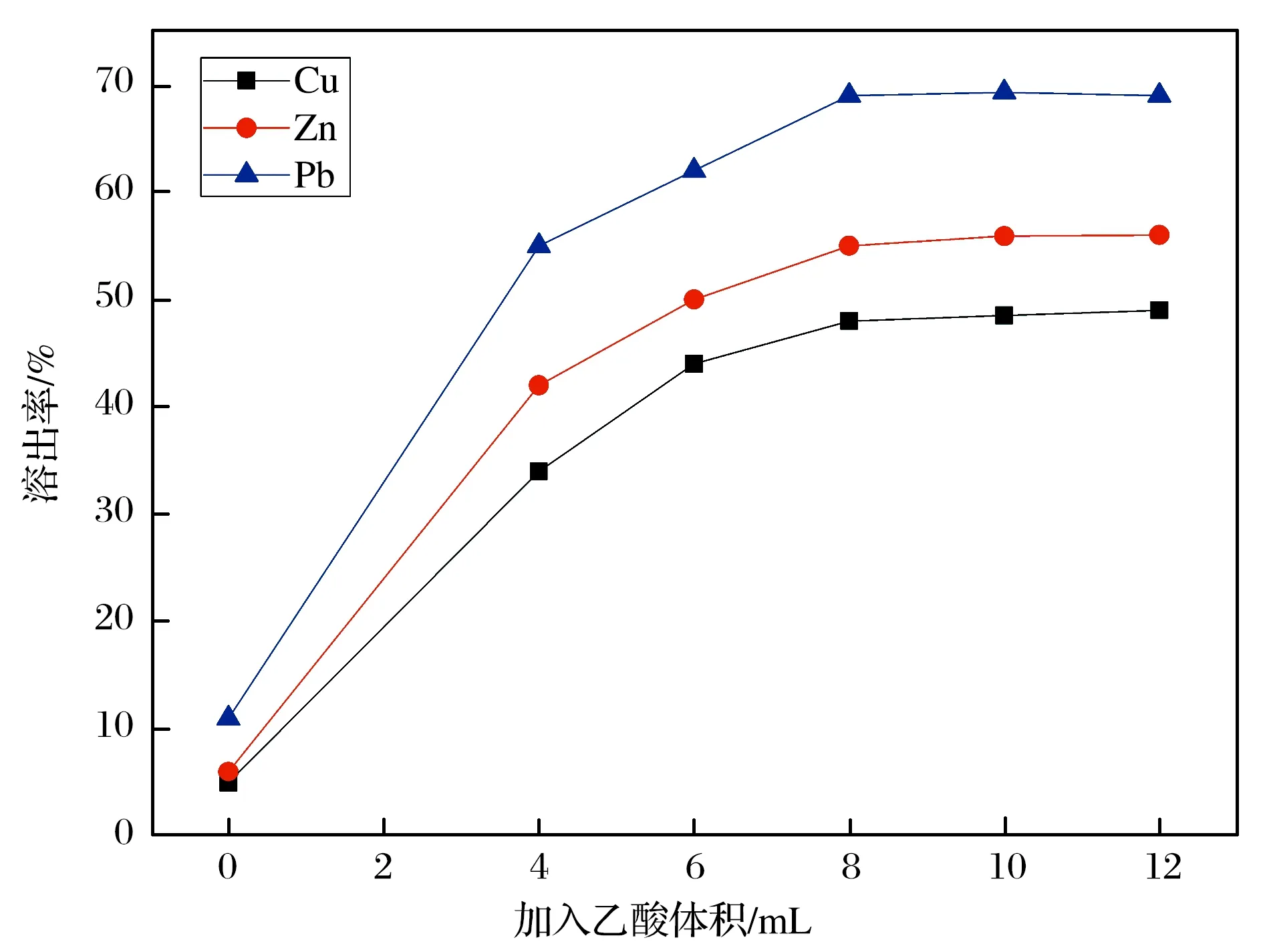
图1 不同体积的乙酸对铜、铅、锌溶出率的影响
2.1.2 样品与熔剂稀释比例的选择
样品与熔剂的配比直接影响样品的熔融程度,充分熔融的样品准确度高、精密度好[11].本文分别以样品与Li2B4O7-LiBO2-LiF混合熔剂的质量比为1∶10、1∶15、1∶20、1∶25、1∶30进行同一样品的制备,各稀释比例下平行制备8个样片,依次上机测定钙的谱线强度并计算标准偏差,同时根据熔融样片的质量选择最佳稀释比,不同稀释比下钙的平均信号强度如图2所示.由图2可见,当稀释倍数小于15时,流动性差,信号波动较大.当稀释倍数大于20时,熔融流动性好,样片清亮,信号强度相对稳定.然而随着稀释倍数的增加,元素的荧光信号强度逐渐降低.通过比较,在稀释比为20时,能够得到相对稳定且具有一定信号强度的结果.因此,本文采用的样品与熔剂稀释比例为1∶20.

图2 不同稀释比对钙谱线强度的影响
2.1.3 脱模剂的选择及用量
熔融制样中,LiBr、NH4I、KBr和NaI是常用的脱模剂.考虑到I-的存在会对某些元素存在干扰,本文选择LiBr作为脱模剂[5].由于LiBr在一定的熔融时间内可挥发,为保证熔融过程具有好的流动性,脱模剂的加入量应严格控制.通过对萤石标准物质GBW07250的多次熔样分析,当加入质量低于0.15 g时,未到熔融终点,样品的流动性就变差,倒模困难.当加入质量高于0.20 g时,倒模较顺利.综合考虑,脱模剂选用0.5 g/mL的LiBr,其加入量为0.5 mL.
2.1.4 熔融温度的选择
对某一萤石样品分别在950、1 000、1 050、1 100、1 150 ℃温度下熔融制样,结果表明,当温度低于1 000 ℃时,熔融物流动性差,样片中含有微小不熔颗粒,样片不均匀,分析结果的精密度差.当温度高于1 050 ℃时,熔融物流动性好,样片清亮.随着温度的升高,脱模剂挥发严重存在倒模困难的现象,因此本文选择1 050 ℃作为熔融温度.此温度下样片质量好,钙的谱线强度相对稳定.
2.1.5 样品量对试验结果的影响
样品经乙酸处理后,部分物质被溶解分离出去导致样品量减少,不同样品减少的量不同.试验发现,如果将剩余样品量与熔剂按1∶20稀释,测定氟化钙的结果普遍偏高.若按照原称样量与熔剂1∶20稀释,则测定样品中的氟化钙偏低.由于无法确定样品量与熔剂比例,因此,样品经过酸处理后不能直接加熔剂进行熔样测定.
本文通过选用某些性质稳定的氧化物如二氧化硅等作为基体补充到原始称样量,可有效避免由于稀释比例的差异带来的影响.考虑到基体干扰问题,分别选用三氧化二铝、二氧化硅、三氧化二铁、氧化镁4种物质作为补加剂,分别对萤石标准样品GBW07251和GBW07253熔融制片测定,测定结果如表2所列.试验结果显示,将灼烧后的样品补加到初始取样量后,测定结果基本满足要求,其中氧化铁的加入使熔融物变得粘稠,二氧化硅的加入测定的氟化钙的值相对较好,因此选择二氧化硅作为补加剂.
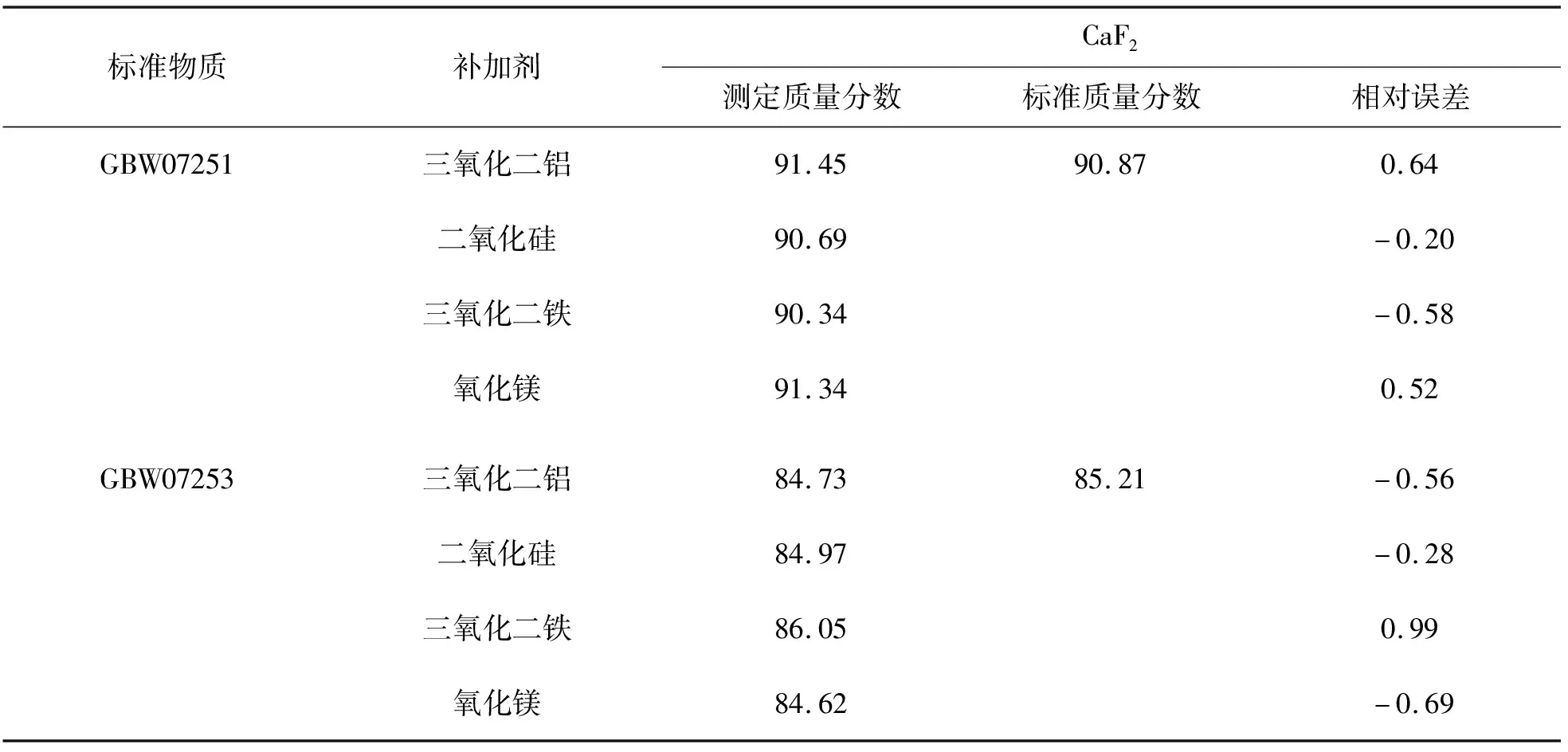
表2 不同补加剂中氟化钙的测定值
2.2 基体校正
熔融制样在很大程度上稀释了样品,可有效地消除了矿物效应以及粒度效应,然而元素间的吸收-增强效应仍然存在.目前在XRF分析中通常采用经验系数法进行校正,本文选用P、Si含量对Ca含量进行校正,修正后的结果明显改善,测定结果如表3所列.相应的计算公式[5]如下:
Wi=(a×I2+b×I+c)×(1+∑dj×Wj)-∑Lj×Wj
j≠I
(1)
式中:Wj—基体元素定量结果;dj—吸收影响系数;Lj—重叠影响系数;Wi—被校正元素的定量结果;I—被校正元素的X射线强度;a,b,c—标准曲线常数.
2.3 精密度
使用标准样品GBW07252和GBW07253分别重复制备12个样片,按照建立的方法进行分析CaF2的含量,测定结果如表4所列.由表4可见,相对标准偏差均小于0.50%,说明该方法具有良好的精密度.
2.4 不同检测方法的结果比对
为了验证该方法的实用性和可靠性,分别对12件萤石样品进行分析,分析结果与容量法比较,测定结果如表5所列.由表5可见,本方法测定CaF2的质量分数与容量法测定的结果相符,表明该方法可靠.
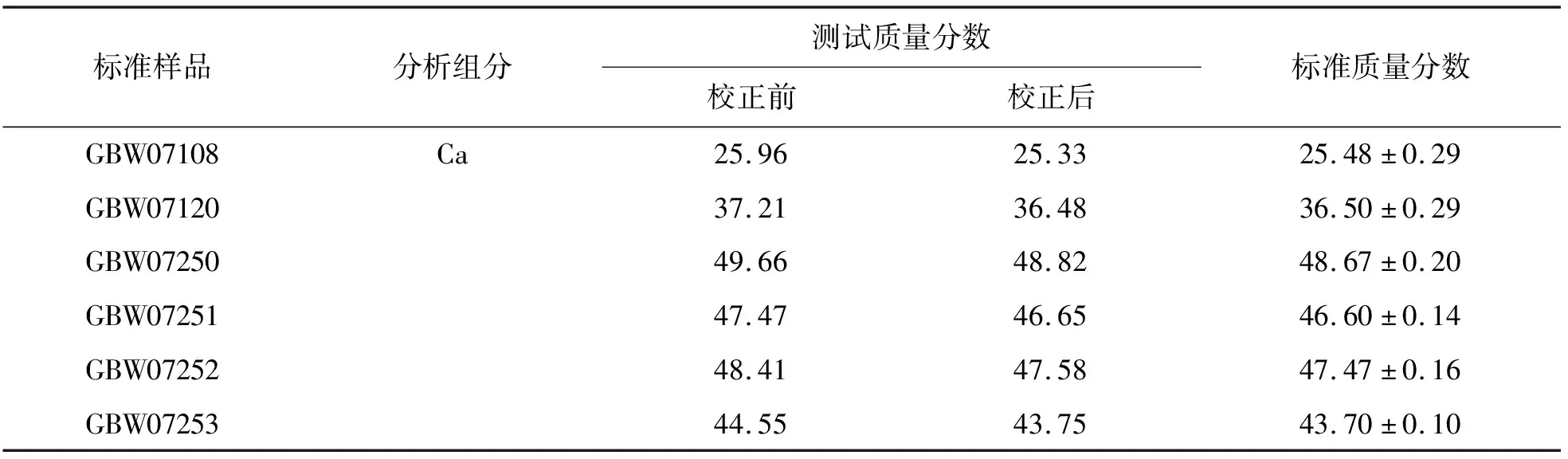
表3 基体校正试验结果

表4 方法精密度
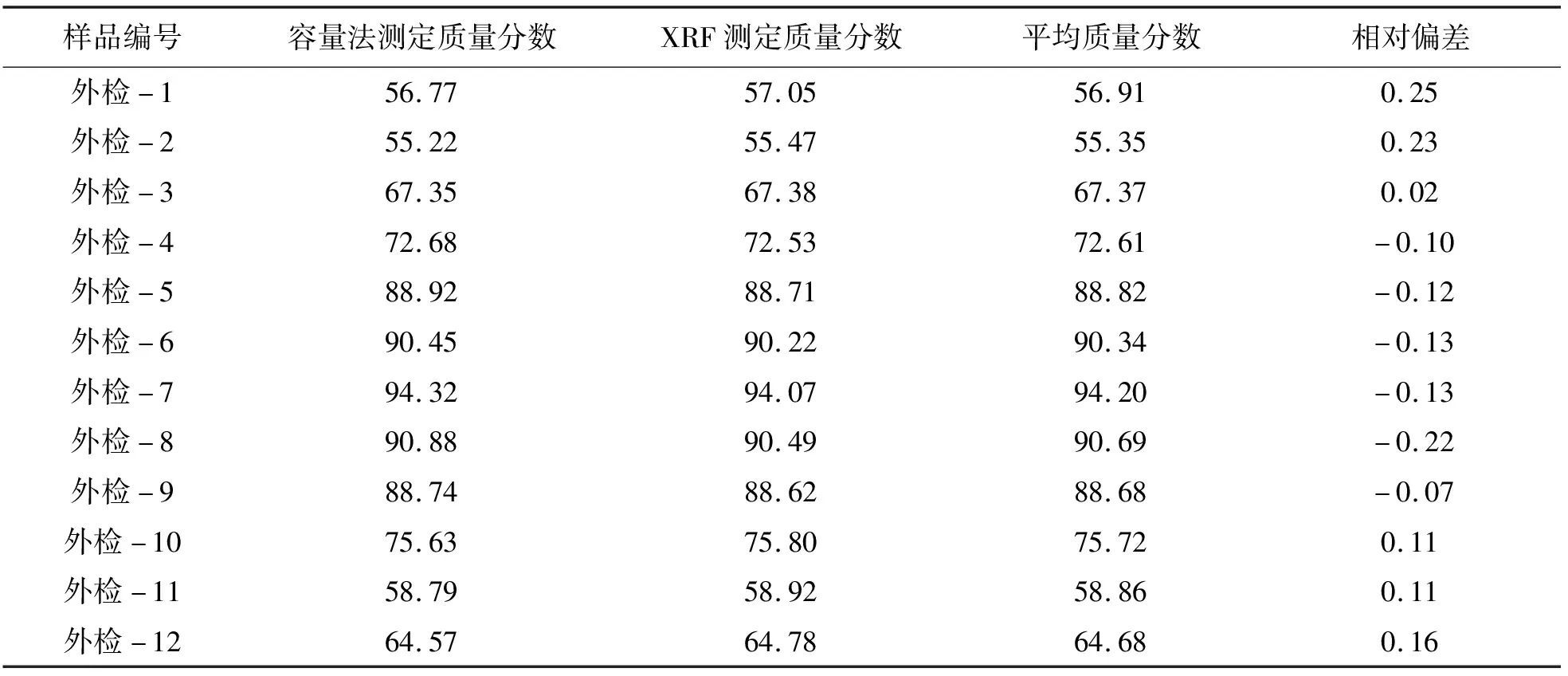
表5 方法准确度
3 结论
传统熔融制样、XRF测定萤石中氟化钙的方法,由于需要前处理导致样品量减少,无法直接测定氟化钙的含量.本文采用光谱纯二氧化硅作为补加剂,较好的解决了这一问题,建立了优化的熔融制样、XRF测定萤石中氟化钙的方法.本方法能较好地解决低含量氟化钙的分析,与EDTA容量法相比,避免了滴定过程中镁等离子的干扰,为萤石分析提供了新的解决思路.
