同位素稀释-超高效液相色谱-串联质谱法测定水产品中孔雀石绿、结晶紫及其代谢物残留量的不确定度评定
2020-04-01柱1徐潇颖1赵超群1陈万勤1梁晶晶1毛思浩1
刘 柱1,徐潇颖1,赵超群1,陈万勤1,梁晶晶1,毛思浩1,华 颖
(1.浙江省食品药品检验研究院,浙江杭州 310052;2.浙江中医药大学药学院,浙江杭州 310053)
孔雀石绿(malachite green,MG)和结晶紫(crystal violet,CV)都属于人工合成的三苯基甲烷类碱性染料,工业上常用于纸张、皮革、纺织工艺品的染色,因具有较强的杀菌、消毒作用且价格低廉,因此被违规用于水产品的养殖和运输过程中,防治鱼、虾等水产品的水霉病、烂鳃病以及寄生虫病等,以延长水产品的存活时间[1-4]。在鱼、虾等水产品中,孔雀石绿和结晶紫常被发现,甚至在鱼苗中亦能检测到[5-6]。孔雀石绿、结晶紫对人类都具有严重的毒副作用,能抑制骨髓造血功能,引起人类的再生障碍性贫血、粒细胞缺乏症、新生儿、早产儿灰色综合征等疾病,低浓度的药物残留也能导致病原菌的耐药性[7-8]。其代谢物-隐性孔雀石绿(leuco malachite green,LMG)和隐性结晶紫(leuco crystal violet,LCV)的毒性则更强[9],因此,世界各国均已禁止在水产品养殖和储运过程中使用孔雀石绿[10],我国2002年农业部公告第235号《动物性食品中兽药最高残留限量》将MG列为禁用药物[11],但近年来依然存在非法使用的问题,给水产品行业和消费者安全带来严重影响[12]。
水产品中MG、CV、LMG、LCV残留量低,基质复杂,前处理和检出技术要求高,目前国内外对水产品中孔雀石绿、结晶紫和其代谢产物的检测方法主要有高效液相色谱法(HPLC)[13-15]和液相色谱-串联质谱法(HPLC-MS/MS)[16-18]。其中HPLC-MS/MS法灵敏度高、选择性好,成为最为常用的方法,但HPLC-MS/MS容易受到基质效应的影响,因此目前常采用同位素稀释法降低质谱的基质效应,提高检测结果的可靠性[19]。有关HPLC法和LC-MS/MS法检测孔雀石绿的不确定评价方法常有报道[20-23],而采用同位素稀释-UPLC-MS/MS法检测水产品中孔雀石绿、结晶紫和其代谢产物的不确定度评定方法的研究报道基本空缺。本研究采用的方法主要依据国家标准GB/T 19857-2005[24]规定的同位素内标LC-MS/MS法,同时为了提高结果的准确性和可靠性,在国家标准方法的基础上增加了D6-结晶紫(D6-CV)和D6-隐色结晶紫(D6-LCV)两个同位素内标,并简化了前处理步骤。
测量不确定度是表征合理地赋予被测量之值的分散性,与测量结果相联系的参数[25],其数值的大小反映了测量结果质量的高低,并直接与检验结果的合格判定相关[26-27]。本实验参考JJF 1059.1-2012《测量不确定度评定与表示》[25]及CNAS-GL006-2019《化学分析中不确定度的评估指南》[28],通过对同位素稀释-超高效液相色谱-串联质谱法检测水产品中孔雀石绿、结晶紫及其代谢物方法的不确定度进行评定,期望为实验室质量控制提供科学、准确、可信的依据,同时为采用同位素内标法测量其他兽药残留量的不确定度评定提供参考。
1 材料与方法
1.1 材料与仪器
孔雀石绿(MG)(纯度≥95%)、结晶紫(CV)(纯度≥92.5%)、隐色孔雀石绿(LMG)(纯度≥98.5%)、隐色结晶紫(LCV)(纯度≥99%) 德国Dr. Ehrenstorfer公司;同位素内标D5-孔雀石绿(D5-MG)(纯度≥99.7%)、D6-结晶紫(D6-CV)(纯度≥99.1%)、D6-隐色孔雀石绿(D6-LMG)(纯度≥100%)、D6-隐色结晶紫(D6-LCV)(纯度≥99.9%) 德国WITEGA公司;实验用水产品(鲈鱼、鲫鱼、明虾) 杭州市当地超市;乙腈、甲醇 色谱醇,德国Merck公司;试验用水 为Milli-pore超纯水;其他试剂 均为国产分析纯。
AB-5500 QTRAP超高效液相色谱-串联质谱仪 配有ESI电喷雾离子源,美国AB SCIEX公司;Shimadzu LC-30AD超高效液相色谱仪 日本岛津公司;Milli-Q Gradient超纯水仪 法国Millipore公司;高速冷冻离心机 美国Thermo Fisher公司;Multi-Prep多样品均质器 美国Pro Scientific公司;全自动氮吹浓缩仪 美国Biotage公司;MS3涡旋混合器 德国IKA公司;XPE-205电子天平 瑞士Mettler Toledo公司;固相萃取装置 美国Waters公司;中性氧化铝固相萃取柱 规格6 mL,500 mg,美国Waters公司。
1.2 实验方法
1.2.1 标准品储备液和稀释液的配制 分别准确称取MG、LMG、CV、LCV各10.0 mg(精确至0.01 mg)至100 mL容量瓶中,用乙腈溶解并定容,即得100 mg/L的标准储备液;用移液枪分别精密移取0.5 mL标准储备液至50 mL容量瓶中,用乙腈定容,即得混合标准工作液a(1000 μg/L);用移液枪精密移取1.0 mL 混合标准工作液a至10 mL容量瓶中,用乙腈定容,即得混合标准工作液b(100 μg/L)。
分别精密称取D5-MG、D6-LMG、D6-CV、D6-LCV各3.0 mg(精确至0.01 mg)至20 mL容量瓶中,用乙腈溶解并定容,即得同位素内标储备液;用可调移液枪分别准确移取各同位素内标储备液溶液0.5~50 mL容量瓶中用乙腈定容,即得1.5 μg/mL的同位素内标工作液。
1.2.2 样品前处理 精密称取5.0 g(精确至0.0001 g)已混匀的样品于50 mL离心管中,加入100 μL同位素内标工作液,再加入8 mL乙腈,超声振荡提取2 min,8000 r/min匀浆提取30 s,然后再4000 r/min离心5 min,转移上清液移至25 mL容量瓶中;用玻棒捣碎离心管中的沉淀,再用8.0 mL乙腈按照上述操作重复提取一次,上清液合并至25 mL容量瓶中,用乙腈定容,摇匀备用。
精确移取2.0 mL样品提取溶液加至已活化(使用前用5 mL乙腈活化)的中性氧化铝柱上,用3 mL乙腈洗脱两次,收集全部流出液和洗脱液于10 mL试管中,35 ℃下氮气吹至近干,加入2.0 mL初始流动相,漩涡溶解复溶,0.22 μm微孔滤膜过滤后转移至样品瓶中用于LC-MS/MS分析。
1.2.3 色谱条件 色谱柱:Waters Atlantis T3(150 mm×2.1 mm,5.0 μm)或相同效能色谱柱;流动相采用10 mmol/L 乙酸铵溶液和乙腈;流速:0.4 mL/min;柱温:30 ℃;进样量:5 μL;梯度洗脱程序见表1。

表1 色谱分离梯度洗脱程序Table 1 Chromatographic gradient elution program
1.2.4 质谱条件 离子源:电喷雾离子源,正离子扫描;扫描方式:多重反应监测;电离电压:5500 V;离子源温度:500 ℃;气帘气流速:30 L/min;碰撞气流量:5 L/min;电源;化合物定性离子对、定量离子对、锥孔电压(DP)、碰撞能量(CE)见表2。

表2 质谱参数Table 2 MS parameters
注:“*”表示定量离子。
在1.2.3和1.2.4的色谱分析条件和质谱分析条件下,MG、LMG、CV、LCV可以获得较好的分离度,如图1和图2所示,表明分析条件合理,可以进行不确定度评定。
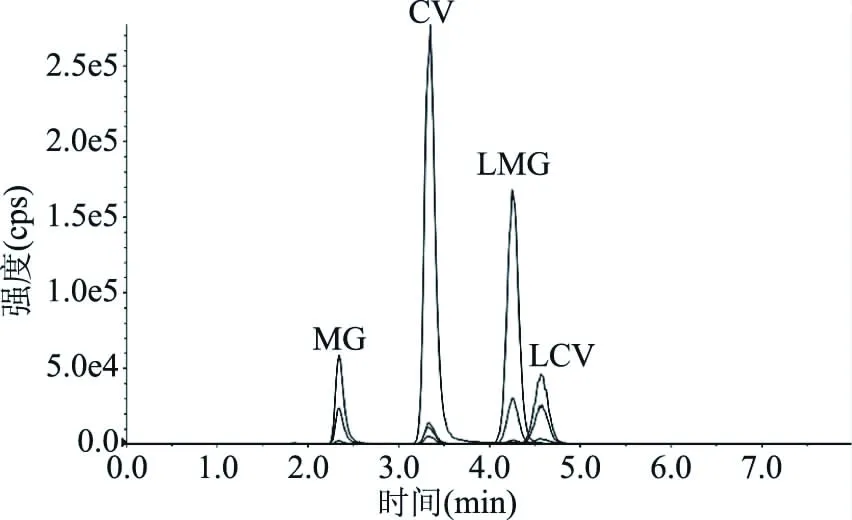
图1 MG、LMG、CV、LCV的提取离子谱图Fig.1 Extracted ion chromalograms of MG,LMG,CV,LCV
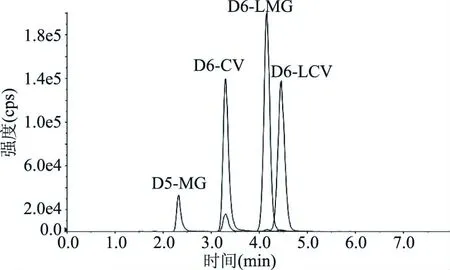
图2 D5-MG、D6-LMG、D6-CV、D6-LCV的提取离子谱图Fig.2 Extracted ion chromalograms of D5-MG,D6-LMG,D6-CV and D6-LCV
1.2.5 数学模型的建立 根据水产品中孔雀石绿、结晶紫及其代谢物残留量的测定原理和方法,待测物中被测物质残留量的计算公式:
式中:X为试样中被测物质的残留量,μg/kg;C为从标准工作曲线得到的试样测定液中被测物质的质量浓度,μg/L;V为试样溶液的定容体积,mL;m为试样溶液代表试样的质量;f为进样重复性、样品预处理过程等因素。
标准工作曲线由最小二乘法进行线性拟合而成,同位素内标法定量,并根据标准工作曲线以峰面积计算得到待测组分的质量浓度。标准溶液配制、体积量取等影响不确定度的相关因素之间采取多元回归分析,确定多个变量的因果关系,建立预测的数学模型。
2 结果与分析
2.1 不确定度来源分析
从测量过程和数学模型,对同位素内标稀释-LC-MS/MS法检测水产品中孔雀石绿、结晶紫及其代谢物残留量的测定结果影响的各种不确定度分离进行分析,具体引入的不确定度来源的因果图如图3所示。
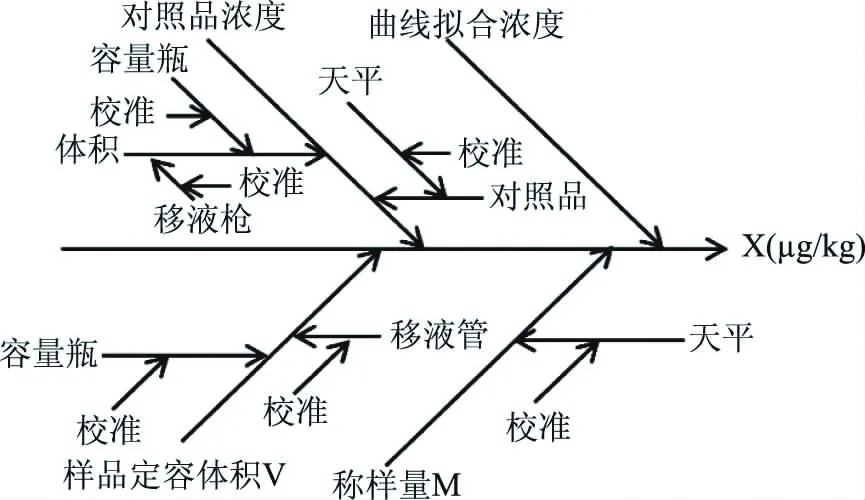
图3 同位素稀释-超高效液相色谱-串联质谱法同时测定水产品中孔雀石绿、结晶紫、隐性孔雀石绿、隐性结晶紫的残留量的不确定度来源Fig.3 Uncertainty source for determination of malachite green, crystal violet,leuco malachite green and leuco crystal violet in aquatic products by isotope dilution-ultra performance liquid chromatography-tandem mass spectrometry
从图3中可以看出,影响测定结果不确定度的主要来源有:a.对照品浓度,配制过程引入的不确定度urel(c);b.样品称量引入的不确定度urel(m);c.样品前处理过程中移取和量取体积引入的不确定urel(V);d.测试过程中曲线拟合以及随机效应引入的不确定度urel(f),包括重复性、回收率、样品均匀性和代表性等因素引入的不确定度。
2.2 待测物引入的不确定度
待测物(C)的不确定度来源由两个因素组成,即标准系列溶液配制和由标准曲线拟合时所产生的不确定度。其中标准系列溶液配制包括储备液配制、稀释及标准曲线溶液配制3个部分[29-31]。
2.2.1 配制标准储备液和稀释液引入的测量不确定度urel(c)(属于B类不确定度评定) 配制标准储备液和稀释液引入的不确定度由以下几个分项构成:a.MG、LMG、CV、LCV四种标准品本身引入的不确定度urel(c1);b.称取标准品引入的不确定度urel(c2);c.10 mL容量瓶定容引入的不确定度urel(c3);d.50 mL容量瓶两次稀释、定容引入的不确定度urel(c4)和urel(c5);e.l mL可调移液器两次移液引入的不确定度urel(c6)和urel(c7);以及配制标准工作曲线引入的不确定度urel(c8)。
由于本方法采用的是同位素内标稀释法,同位素内标浓度不参与测量结果的计算,因此在此过程中可不考虑同位素内标配制的不确定度,只评估在绘制标准工作曲线时加入同位素内标时引入的不确定度。
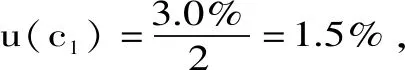

表3 标准品系列溶液配制过程中引入的不确定度Table 3 Uncertainty resulting from standard solution preparation
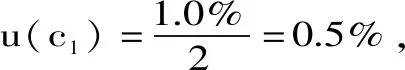



2.2.1.3 10 mL容量瓶定容引入的不确定度urel(c3) 本实验所用10 mL容量瓶(A级)在20 ℃条件下的允差Δ为±0.02 mL,取矩形分布,则由定容引入的不确定度为:
2.2.1.4 50 mL容量瓶两次稀释定容引入的不确定度urel(c4)、urel(c5) 本实验所用50 mL容量瓶(A级)在20 ℃条件下的允差Δ为±0.05 mL,取矩形分布,则由定容引入的不确定度为:

2.2.1.6 配制标准系列引入的不确定度urel(c8)(属于B类不确定度评定) 分别使用200和1000 μL的移液器,分别精密移取混合标准工作液b(100 μg/L)50、100、200、400、600、800、1000 μL,置于10 mL容量瓶中,再使用50 μL的移液器向每个容量瓶中加入40 μL内标工作液,最终用初始流动相定容即得标准系列。按照JJG 196-2006《常用玻璃量器检定规程》[33]和JJG 646-2006《移液器检定规程》的要求,配制标准系列过程中所使用的玻璃量器和移液器引入的相对标准不确定度见表3。
则由标准系列溶液配制过程中引入的相对不确定度为:

表4 标准曲线数据及处理Table 4 Standard curve data and processing
通过研究配制标准系列溶液过程中的不确定度评定发现,相对于非同位素内标法而言,不确定度的来源中增加了因添加同位素内标时引入的不确定度,因此应用同位素内标法进行检测时,应将同位素内标稀释至合适的浓度,以便在标准系列工作液配制时添加适当的同位素内标溶液体积,降低因配制标准系列溶液引入的不确定度。
2.2.2 采用最小二乘法拟合标准工作曲线求得试样浓度过程引入的不确定度urel(Cpred)(属于B类评定) 对“2.2.1.6”配制的7个水平的标准系列工作液,其对应的浓度为0.5、1、2、4、6、8、10 μg/L,按照“1.2.3”和“1.2.4”的方法重复测定2次,测得4种化合物的峰面积和相应同位素内标峰面积,结果如表4所示,以标准溶液浓度(μg/L)为横坐标,以化合物和相应同位素内标峰面积比值为纵坐标,得到MG、LMG、CV和LCV的回归方程、截距和线性决定系数R2结果如表5所示。
各化合物拟合而成的线性回归方程为Y=aX+b(b为截距,a为斜率),对购买的阳性样品中各待测化合物进行6次重复测定,其结果见表5,根据样品中各待测化合物与对应同位素内标的比值,利用标准工作曲线的线性回归方程求得相应的浓度,根据贝塞尔公式,标准曲线的拟合标准偏差为:
式中:S(A)为标准曲线的拟合标准偏差;Ai为标准溶液与内标溶液响应值的比值;Ci为标准系列溶液的浓度,μg/L;n为标准溶液测定的总次数(不包含0点);a为标准曲线的斜率;b为标准曲线的截距。
将表4和表5中的数据带入S(A)计算公式,得出MG的S(A)=0.0181091;LMG的S(A)=0.00724365;CV的S(A)=0.0118181;LCV的S(A)=0.00332523。
则由最小二乘法拟合标准工作曲线所引入的标准不确定度的计算式为:
将相应的数据带入公式进行计算,得出MG的u(Cpred)=0.055184;LMG的u(Cpred)=0.043917;CV的u(Cpred)=0.0308395;LCV的u(Cpred)=0.0472514。
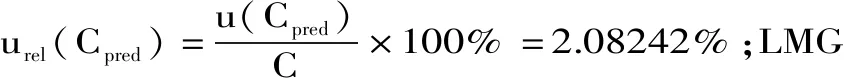
2.3 样品前处理过程引入的不确定度

2.3.2 添加内标溶液引入的不确定度urel(I)(属于B类不确定度评定) 采用200 μL移液器移取100 μL混合同位素内标工作液(1.5 μg/mL)至称取的样品中导致的不确定度urel(I)=urel(V200)=0.866%。
由于加入内标后,样品中MG、LMG、CV和LCV与其对应的同位素内标D5-MG、D6-LMG、D6-CV、D6-LCV含量的比值不会因为体积的变化而变化,因此在样品的前处理过程中提取液的定容体积、移取待净化液的体积和净化浓缩后的定容体积都对以化合物和相应同位素内标含量的比值没有影响,因此这些过程对检测结果的不确定度没有影响。
2.4 回收率产生的不确定度urel(R)
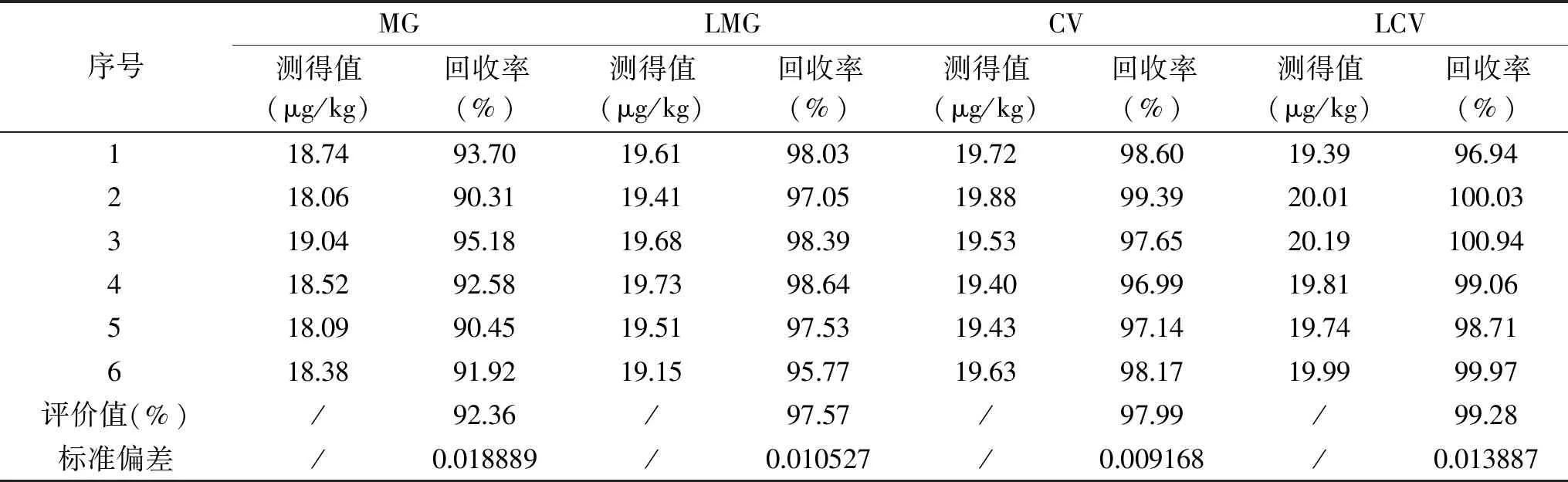
表6 重复测定检测结果Table 6 Repeatability of determination of MG,LMG,CV and LCV
注:“/”表示未进行计算。
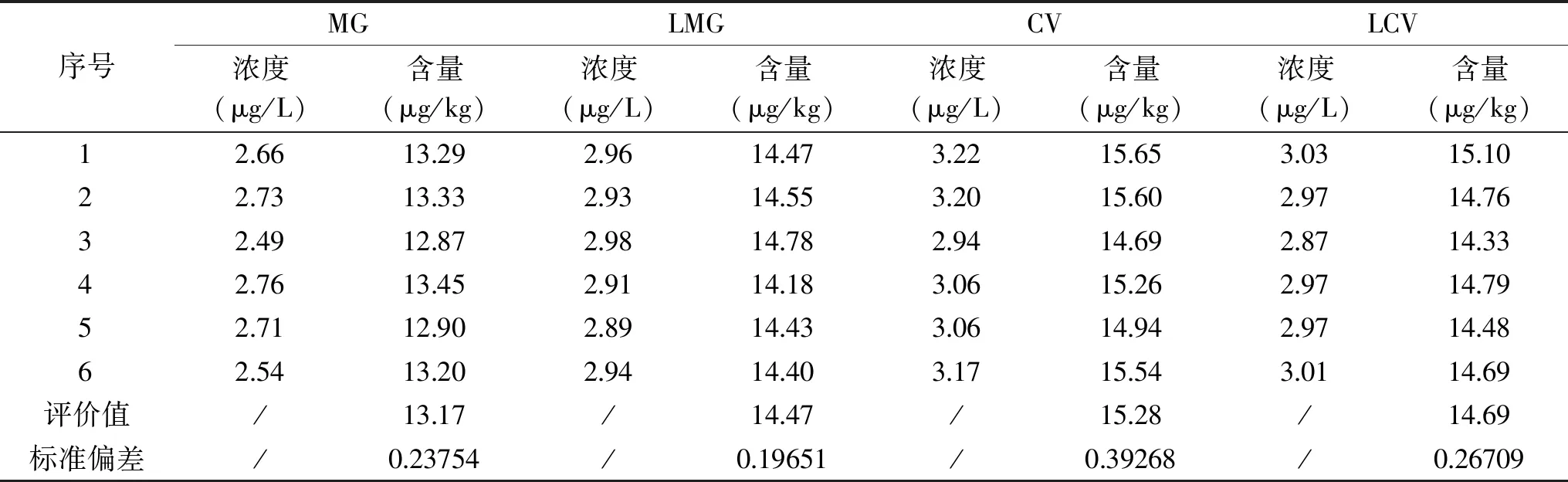
表7 重复测定检测结果Table 7 Repeatability of determination of MG,LMG,CV and LCV
在6份空白鱼肉样品中添加MG、LMG、CV、LCV混合标准工作液,使样品中孔雀石绿、结晶紫及其代谢物的浓度为20 μg/kg,按照“1.2.2”进行样品前处理,再按照“1.2.3”和“1.2.4”的方法利用同位素稀释法进行检测,测得加标回收的结果详见表6。

2.5 测试重复性引入的不确定度urel(f)
测试过程随机效应引入的不确定度,包括样品预处理过程、样品经中性氧化铝柱净化过程、进样重复性、样品均匀性和代表性等影响因素,属于A类不确定度评定。根据“2.2.2”表5中阳性样品各待测化合物6次重复测定的结果计算该阳性样品中MG、LMG、CV和LCV的含量见表7。
2.6 测量不确定度的评定与报告


计算得到MG、LMG、CV和LCV的相对合成标准不确定度详见表8。

表8 MG,LMG,CV 和 LCV的相对标准不确定度Table 8 List of relative uncertainty components for MG,LMG,CV and LCV

表9 不确定度评估结果Table 9 Uncertainty evaluation for the determination
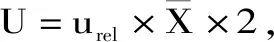
3 结论
采用多种同位素稀释-超高效液相色谱-串联质谱法(UHPLC-MS/MS)同时测定水产品中孔雀石绿、结晶紫、隐性孔雀石绿、隐性结晶紫残留量的实验过程中,称量、提取、净化和质谱测定等过程均会引入不确定度,本研究系统深入地对样品的称量、玻璃仪器、标准溶液的配制和稀释、样品重复性测定、加标回收、标准曲线拟合等因素进行综合考量,结果表明,在样品的前处理过程中提取液的定容体积、移取待净化液的体积和净化浓缩后的定容体积都对以化合物和相应同位素内标含量的比值没有影响,因此这些过程对检测结果的不确定度没有影响。标准溶液配制和标准曲线拟合过程引入的不确定度最大,其次是重复性,而标准溶液配制中又以标准曲线配制引入的不确定度最大,而样品称量引入的不确定度可忽略不计。因此,在实际操作过程中,可通过调整同位素内标的加标体积,增加混合标准系列溶液的测定次数,增加平行样品测定,保持超高效液相色谱-串联质谱仪器较高的灵敏度,并定期对所涉仪器进行检定和提高操作人员的熟练水平,来减小测量不确定度,从而保证检测结果的准确性。
