Be4N3-:一个含超短铍-铍间距的超卤素负离子
2020-04-01仝文彦赵涛涛吴艳波
仝文彦,赵涛涛,吴艳波
(山西大学 分子科学研究所,山西 太原 030006)
0 引言
金属通常具有较大的原子半径,这导致分子中的金属-金属原子间距较大。因此,如果有分子的金属-金属原子间距变得异乎寻常的短,那么该分子会受到广泛的关注,因为这表明两个金属原子间发生了直接或间接的强烈相互作用。当金属原子间距小于1.900 Å时,人们一般称之为“超短金属-金属间距(Ultrashort metal-metal distance, USMMD)”[1-2]。起初,USMMD是在过渡金属原子间被发现的,这是因为过渡金属的壳层电子结构允许金属原子间形成五重键,这使相应的原子间距得到极大地缩短。目前,在晶体结构中,Cr-Cr五重键可以短至1.706 Å[3-10],而在理论预测的分子中,Cr-Cr间距可以被缩短至1.650 Å[11-14]。
然而,对于主族金属来说,在它们之间形成USMMD(即用主族金属来支撑USMMD)有很大的困难。首先,主族元素的壳层电子结构使得主族元素间最高只能形成同核三重键;其次,对主族金属来说甚至形成同核三重键都比较困难,因为s区碱金属和碱土金属的价电子数不足,而p区元素的最外层s孤对电子呈现出了很高的惰性[15]。但在这方面,铍由于具有最小的金属半径并且具有典型的缺电子性而成为主族金属中的特例。我们课题组前期的研究发现,虽然铍靠自身的电子无法形成三重键而支撑USMMD,但在合适桥位原子的帮助下,两个铍原子间可以通过形成多个成键分子轨道、增强静电吸引或将这两种策略结合而达到大幅缩短铍-铍间距的目的[16-19]。目前,最短的铍-铍间距是1.627 Å[18],比最短的过渡金属间距(1.650 Å)[13]还短。作为对比,经典的铍-铍双键键长在1.950 Å左右[20]。
过渡金属五重键目前已经在合成化学中有了较多应用,但超短铍-铍间距的应用报道较少。我们课题组之前的研究发现[E→Be2H3←E]+(E=NH3, PH3, Ar, Kr, Xe)[19]是含有超短铍-铍间距的超碱金属[21]离子。超碱金属是指“垂直电离能(VDE)小于3.89 eV(周期表中VDE最小的铯原子的数值)的超原子”,它是超原子的一个重要类型,通常具有超强的还原性,在氧化还原反应研究中可能有重要应用。超原子的另一个重要类型是超卤素[22],它是指“电子亲和能(EA)大于3.62 eV(周期表中EA最大的氯原子的数值)的超原子”,通常具有超强的氧化性,同样在氧化还原反应研究中可能有重要应用。与碱金属或卤素类似,超碱金属或超卤素通常以其正离子或负离子的形式存在并表现出很高的稳定性。相应的,人们一般通过对比超原子正离子与Cs+离子的EA来判断它是否属于超碱金属正离子,通过对比超原子负离子与Cl-的VDE来判断它是否属于超卤素负离子。值得注意的是,实验中测量Cs+离子的EA是通过测量Cs原子的VDE来实现的,Cl原子的EA也是通过测量Cl-的VDE来实现的。
由于具有USMMD的分子中的金属原子间都有直接或间接的较多相互作用,通常会使分子的还原性较强而氧化性较弱,因此,目前尚无含有USMMD的超卤素报道,而设计含有USMMD的超卤素是含有非经典成键结构设计领域的一个较大挑战。在本文的工作中,我们设计了带负电荷的含有USMMD的Be4N3-分子并发现其VDE高达3.70 eV,这符合超卤素负离子的判断标准,从而首次从理论上预测了含有USMMD的超卤素负离子。
1 计算方法
对C3vBe4N3-分子(1a),我们在B3LYP/aug-cc-pVTZ和MP2/aug-cc-pVTZ水平进行了几何构型优化和频率分析,得到了相似的优化结构和振动频率结果。我们又在CCSD(T)/aug-cc-pVTZ水平下对其结构进行了进一步优化,发现B3LYP、MP2和CCSD(T)三种方法优化的结构没有显著差别。同时,我们在B3LYP/aug-cc-pVTZ水平下对1a的波函数稳定性进行了分析。为了理解分子中的成键,我们分别在B3LYP/aug-cc-pVTZ和B3LYP/6-31G(d)水平下进行了自然键轨道(NBO)[23]和适应性自然密度划分(AdNDP)[24]分析。分子得失电子的难易程度使用垂直电离能(VDE)和电子亲和能(EA)来描述,相关数据使用外价层格林函数法(OVGF)[25],在OVGF/aug-cc-pVTZ水平下进行计算。为了探索该分子的热力学稳定性,我们使用随机搜索算法[26-27]对组分Be4N3-的势能面进行了搜索:随机生成的初始结构首先过滤掉极不合理的结构,然后将剩余的结构在B3LYP/6-31G(d)水平下进行优化,接着选取能量最低的10个结构在B3LYP/aug-cc-pVTZ水平下进一步进行几何构型优化和频率分析计算;随后在CCSD(T)/aug-cc-pVTZ水平下对B3LYP/aug-cc-pVTZ的优化结构进行单点能计算;最后异构体的相对能量在CCSD(T)/aug-cc-pVTZ单点能加B3LYP/aug-cc-pVTZ吉布斯自由能校正的水平下进行比较。为了研究该分子的动力学稳定性,我们在B3LYP/6-31G(d)水平和4、298和500 K下进行了50 ps Born-Oppenheimer分子动力学(BOMD)[28-29]模拟,动力学步长约为1.5 fs,模拟中的结构演变用相对于优化结构的均方根偏差(RMSD)来表示。随机搜索算法通过GXYZ 2.0[30-31]程序实现,AdNDP分析使用AdNDP程序[32]进行分析,CCSD(T)的计算使用MolPro 2012.1软件[33]运行,而其他计算使用Gaussian 09程序[34]运行。
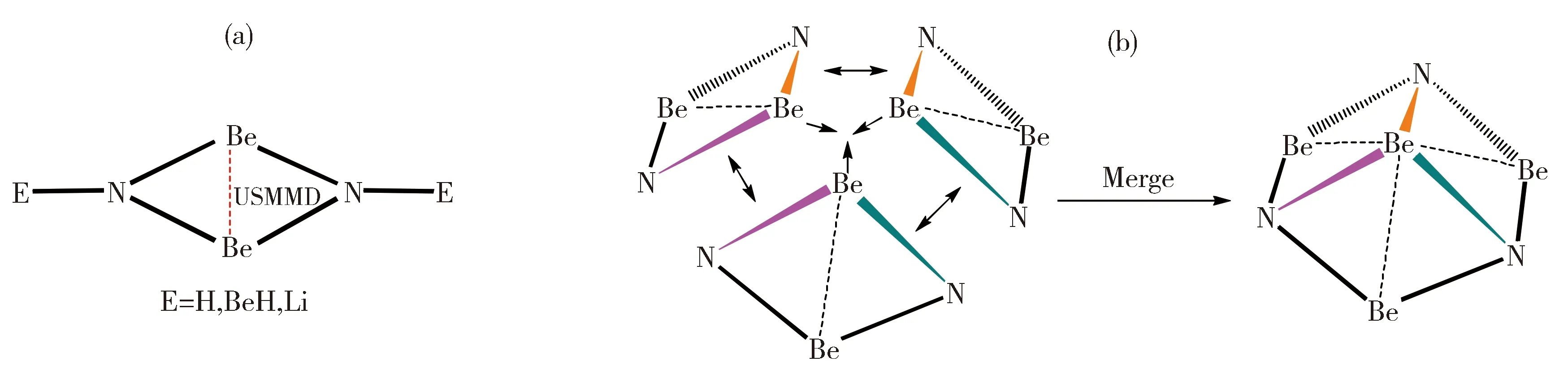
图1 Be2N2E2的结构示意图(a)及本文用到的设计思想(b)Fig.1 The structures of Be2N2E2 species (a) and the designing strategy used in this work (b)
2 结果与讨论
2.1 设计Be4N3-负离子
我们课题组之前报道的Be2N2E2(E=H, BeH,Li,[16]见图1-a)系列分子虽然Be-Be间距很短,但它们都是三元体系,这增加了实验合成的难度。由于这些分子都有一个菱形Be2N2核心结构,我们直接用该结构作为单元,通过边-边融合的方式组装成Be4N3结构(图1-b),该结构在中性时有23个骨架电子。很意外的是,这个结构与我们课题组近期报道的含USMMD的D3hBe5N3+ [18]相比仅少了一个轴向的铍原子。由于D3hBe5N3+是一个具有24个骨架电子的全局极小结构,我们为Be4N3分子添加了一个电子使其骨架电子数也为24,从而得到了C3vBe4N3-负离子(1a)。在B3LYP/aug-cc-pVTZ水平下,1a是波函数稳定的能量极小结构,MP2/aug-cc-pVTZ水平确认了1a是能量极小结构,其CCSD(T)/aug-cc-pVTZ水平下的优化结构见图2。如图所示,1a的中心Be原子与外围3个Be原子间的距离均为1.853 Å,符合USMMD的判据,可被称为超短铍-铍间距。虽然该间距比此前报道多数超短铍-铍间距要长,但该分子首次在一个分子中形成了三个超短铍-铍间距。该分子的中心Be原子与N原子间的距离为1.731 Å,外围Be原子与N原子间的距离为1.562 Å,分别代表了分子中一类较弱和一类较强的N-Be相互作用。
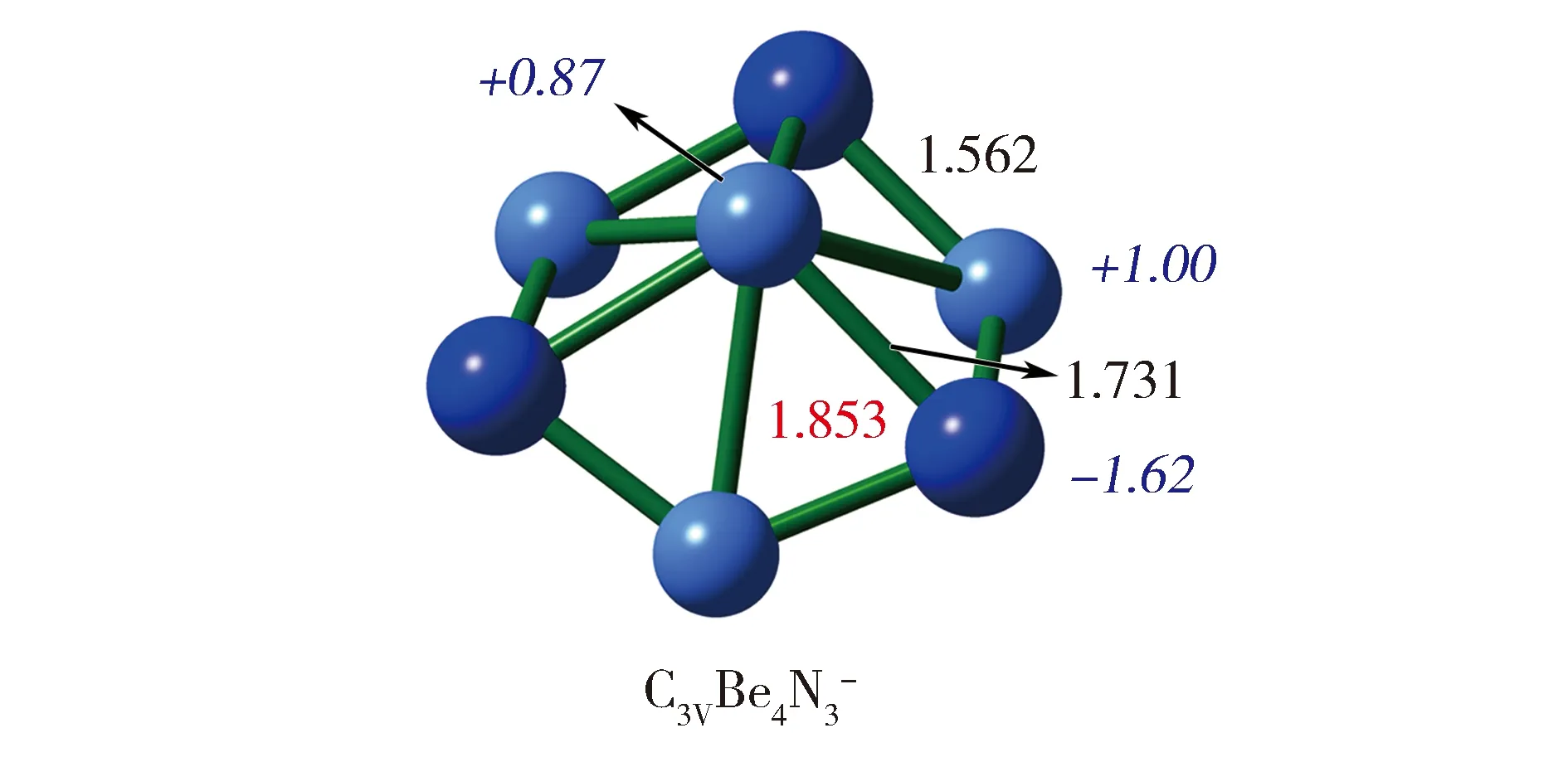
图2 在CCSD(T)/aug-cc-pVTZ水平优化的Be4N3-。Be-Be间距(Å)、其他原子间距离(Å)和自然电荷(|e|)分别用红色字、黑色字和蓝色斜体字标示。Fig.2 CCSD(T)/aug-cc-pVTZ-optimzied structure of C3v Be4N3-.The Be-Be distances (Å), other interatomic distances (Å),and the natural charges (|e|) are shown in red,black, and italic blue fonts, respectively.
2.2 稳定性分析
在B3LYP/aug-cc-pVTZ的水平下,1a的HOMO-LUMO能隙为2.98 eV,表明分子具有较好的电子结构稳定性。OVGF/aug-cc-pVTZ计算得到的VDE为3.70 eV,高于Cl-的VDE,因此1a可被称为超碱金属负离子,同时表明它很难失去电子。OVGF计算还表明该分子的VEA为+2.33 eV,因此该分子也没有得到电子的趋势。从得失电子的角度来看,1a应该具有较好的稳定性。值得一提的是,该分子是首个具有USMMD的超卤素负离子。
为了进一步确认1a分子的稳定性,我们使用随机搜索算法[26-27]搜索了该分子所属的Be4N3-势能面。经过初始优化、精确优化和高精度能量计算,我们发现1a是势能面上的全局能量极小结构。能量次低结构(1b,见图3)的基态是三重态,比1a在能量上高23.6 kcal/mol,因此1a具有良好的热力学稳定性。同时,我们还使用BOMD[28-29]在4、298和500 K下分别作了50 ps分子动力学模拟。模拟过程中的RMSD变化见图4。如图所示,在1a的模拟中RMSD数值变化比较平稳,没有出现向上的跳跃,RMSD的绝对值和波动幅度也很小,这说明1a在动力学模拟中可以保持其基本结构,表现出了较好地动力学稳定性。作为一个动力学稳定的全局能量极小结构,1a很有可能在负离子光电子能谱实验中被合成和表征。
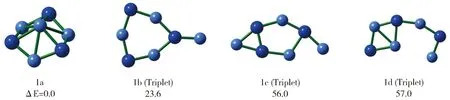
图3 Be4N3-势能面上能量最低的四个异构体及其相对能量(1a的能量设为0)Fig.3 The structures of four lowest isomers on Be4N3-potential energy surfaces and their relative energies (that of 1a is set as zero)
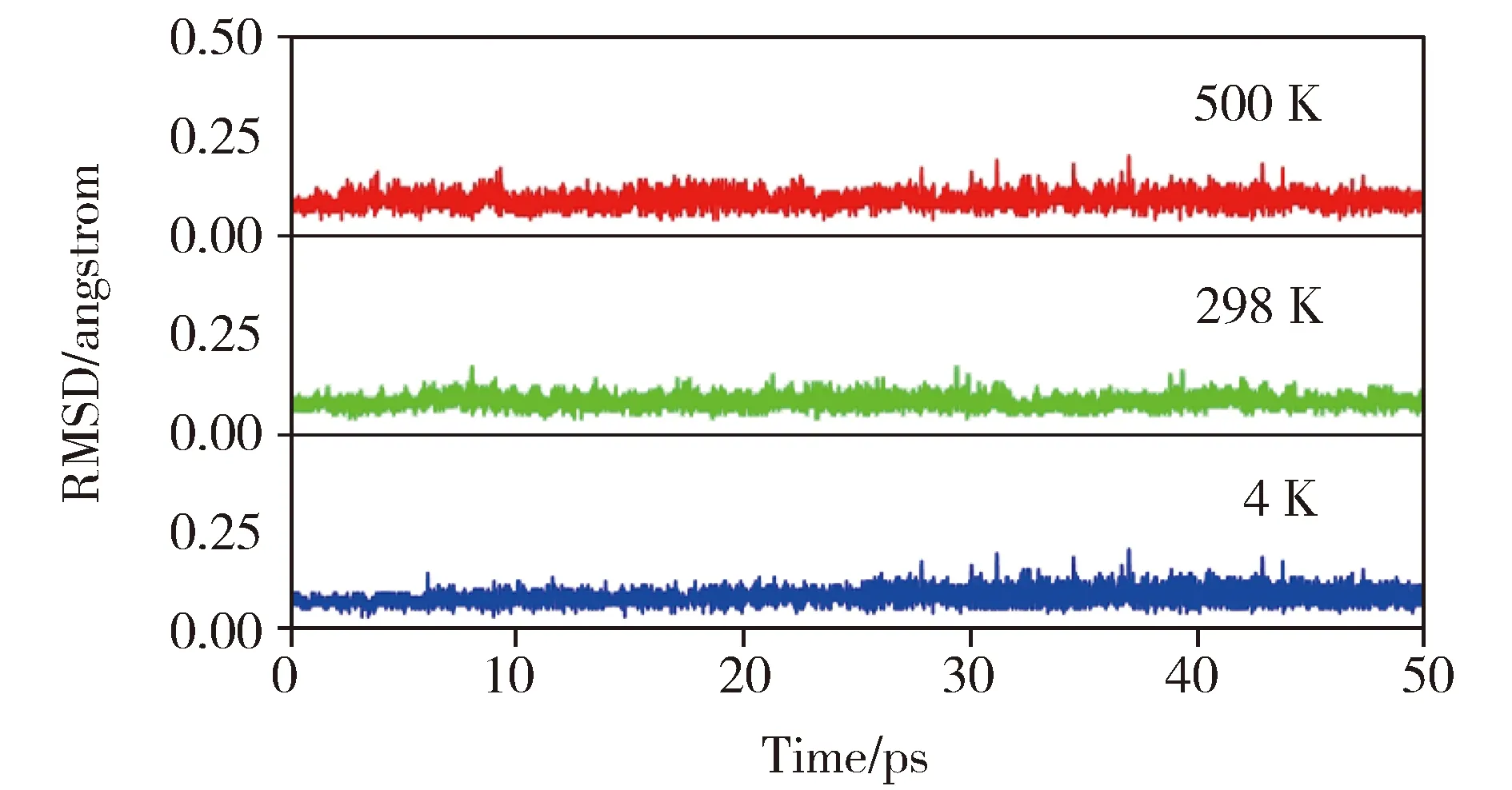
图4 298和500 K下对1a的BOMD模拟中RMSD随时间的变化图Fig.4 Plots of RMSD versus simulation time for BOMD simulations of 1a at 4, 298, and 500 K, respectively
2.3 电子结构分析
为了阐明分子中Be-Be间距能被缩短到1.853 Å的原因,我们对该分子作了电子结构分析。我们首先使用AdNDP[24]方法对该分子做了成键分析。作为传统NBO[23]方法的拓展,AdNDP可以通过nc-2e键(n取值可以从1到分子内的总原子数)的方式反映的成键特点。一般来说,在轨道占据数合理的前提下,n取值越小越好。如图5所示,在1a的12对价电子中,3对是N原子的孤对电子,6对用于形成分子外周骨架的2c-2e N-Be键,另外的3对用于形成中心Be原子与外围N原子间的2c-2e键。这样,每个Be2N2单元由4个N-Be 2c-2e键围绕,而单元内的2个Be原子间没有显著的成键轨道,这与Be2N2E2(E=H, BeH, Li)的Be2N2菱形核心的成键模式是类似的。

图5 1a的AdNDP成键模式及每个轨道的电子占据值(ONs)Fig.5 AdNDP view of chemical bonding in 1a.The occupation numbers (ONs) are indicated
同时,该分子的NBO电荷也与Be2N2E2(E=H, BeH, Li)[16]中Be2N2菱形核心较为相似,1a的每个Be2N2菱形包含1个中心Be原子、1个外围Be原子和2个外围N原子,如图2所示,中心Be原子和外围Be原子自然电荷分别为+0.87 |e|和+1.00 |e|,N原子自然的电荷为-1.62 |e|,因此,每个Be2N2菱形中的Be原子和N原子存在强库伦吸引作用,加上N-Be间还存在的2c-2e键,Be2N2菱形单元中在Be-N间的共价作用和静电作用极大地拉近了N-Be间距。同时由于N原子都带有较大负电荷,有较强的静电排斥,N原子倾向于相互远离。在N-Be间强烈吸引作用和N-N间强烈的排斥作用共同作用下,2个Be原子间距被缩短到了USMMD范围之内。这也表明,1a是含有超短Be-Be间距而Be原子间没有键的分子家族的一个新成员。
3 结论
通过对C3vBe4N3-结构的设计证明了在含有超短间距的分子中可以同时实现具有超卤素负离子属性。这个分子的中的Be-Be间距只有1.853 Å,符合USMMD的判据;其VDE高达3.70 eV,又符合超卤素负离子的判据。它是第一种具有USMMD的超卤素负离子。分子虽然含有超短Be-Be间距,但不是通过形成Be-Be多重键而是通过强烈的N-Be吸引以及N-N排斥作用实现的。该分子具有良好的电子结构、热力学稳定性和动力学稳定性,适合使用光电子能谱方法进行气相合成与表征。我们期望有实验工作者能去探索这个有趣的结构,推进超原子和金属-金属键领域的发展。
