氟喹诺酮混合溶液标准物质研制
2020-03-26周剑张丽媛王敏杨梦瑞王彤彤
周剑,张丽媛,王敏,杨梦瑞,王彤彤
(中国农业科学院农业质量标准与检测技术研究所,农业农村部农产品质量安全重点实验室,北京 100081)
恩诺沙星(ENR)、环丙沙星(CIP)、沙拉沙星(SAR)、达氟沙星(DAN)、氧氟沙星(OFL)、培氟沙星(PFL)、诺氟沙星(NFX)、洛美沙星(LFL)属于氟喹诺酮类药物(FQs),是喹诺酮药物发展到第三代的产物,具有杀菌效果好、抗菌谱广、机体吸收快等优点[1],广泛应用于预防和治疗动物疾病[2–3]。但使用过量或者使用不当会导致动物源性食品中药物残留[4],如进入人体易造成人体内分泌系统、消化系统、生殖系统等的损伤[5–6],并产生细菌耐药性[7–9]。因此欧盟、北美等国均规定FQs 在动物源食品中的最高残留限量不得超过10~400 μg/kg[10]。我国农业农村部也发布了《兽药停药期规定》和《动物源性食品中兽药最高残留限量》,以规范FQs 的使用[11]。
目前喹诺酮类药物残留的检测方法主要有微生物法、理化检测法和免疫检测法等[11–12]。其中理化检测法主要有电化学法[13]、高效液相色谱法[14–15]和液相色谱串联质谱法[16–17]等。农业农村部也公布了基于液相色谱串联质谱的方法标准用于例行监测工作,但检测结果的准确性、可比性、可溯源性需要配套相应的标准物质才能得以保证[18]。
目前由农业农村部农产品质量标准研究中心研制的恩诺沙星、环丙沙星、沙拉沙星、氧氟沙星、诺氟沙星和洛美沙星溶液标准物质均为单一成分标准溶液;而达氟沙星只有纯度标准物质暂无溶液标准物质;培氟沙星还没有相关标准物质。这些单一成分标准物质纯品或溶液,在日常使用时,需要进一步稀释为标准储备溶液,再根据实验需要稀释成工作溶液。对于多种药物同时检测,还要配制成混合标准工作溶液。纯品标准物质价格昂贵,每次实验实际用量很小,造成极大浪费;同时,实验室自行配制储备液,不仅延长实验准备时间,还会引入不确定的误差,影响实验结果。因此研制均匀、稳定性良好、可溯源的氟喹诺酮类混合溶液标准物质,对于保证检测结果准确可靠、节省实验时间和经济成本,以及对国家农产品质量安全例行监测溯源体系的建立具有重要意义。
1 实验部分
1.1 主要仪器与试剂
高效液相色谱仪:LC-20AD 型,日本岛津公司;
质谱仪:Triple QuadTM3500 型,美国应用生物系统公司;
核磁共振仪:Avance III 型,德国布鲁克公司;
卡尔费休水分滴定仪:TOLEDO DL32 型,瑞士梅特勒–托利多公司;
分析天平:XS105 型,感量为0.01 mg,瑞士梅特勒–托利多公司;
电子天平:UMX2 型,感量为0.001 mg,瑞士梅特勒–托利多公司;
电感耦合等离子体质谱仪:X series 2 型,美国赛默飞世尔科技公司;
7 种氟喹诺酮药物纯度标准物质:基本信息见表1。

表1 7 种氟喹诺酮类药物纯度标准物质
培氟沙星:纯度为92.19%,不确定度为1.00%,德国Dr. Ehrenstorfer 公司;
氘代甲醇:纯度为99.6%,美国西格玛奥德里奇公司;
乙腈、甲醇:色谱纯,德国默克公司。
1.2 仪器工作条件
1.2.1 液相色谱仪
(1)测定培氟沙星原料的主成分纯度。色谱柱:Agilent Zorbax SB-Aq 柱(250 mm×4.6 mm,5.0 μm,美国安捷伦科技公司);流动相:流动相A 为20 mmol/L 醋酸铵+0.1%甲酸水溶液,流动相B 为乙腈,(流动相A 与流动相B 的体积比为45∶55);洗脱方式:等度洗脱;流量:1 mL/min;进样体积:10 μL;紫外检测器波长:279 nm。
(2)测定8 种氟喹诺酮药物混合溶液。色谱柱:Agilent Zorbax SB-Aq 柱(250 mm×4.6 mm,5.0 μm,美国安捷伦科技公司);流动相:流动相A 为20 mmol/L 醋酸铵+0.1%甲酸水溶液,流动相B 为乙腈,流动相C 为甲醇;流量:1 mL/min;进样体积:10 μL;紫外检测器波长:290 nm;梯度洗脱程序见表2。

表2 梯度洗脱程序
1.2.2 质谱仪
离子源:电喷雾离子源(ESI);离子源电压:4 500 V;离子源温度:550 ℃;气帘气压力:172 kPa;碰撞气压力:55 kPa;雾化气压力:310 kPa;辅助气压力:310 kPa;扫描范围:50~340 Da。
1.2.3 核磁氢谱仪
探头温度:20.25℃;扫描宽度:8 223.43 Hz;激发脉冲角度:30 度;脉冲序列:zg30;弛豫延迟时间:60 s;采样时间:3.98 s;累计采样次数:128 次;偏置频率:2 464.51 Hz;接收增益:64.00;谱宽:20.00×10–6;纵向弛豫时间:8.5 s。
1.2.4 电感耦合等离子体质谱仪
射频功率:1 450 W;雾化室温度:2℃;等离子体气:氩气,流量为16 L/min;载气:氩气,流量为1.23 L/min;蠕动泵转速:0.4 r/s;采样深度:7.8 mm;同位素驻留时间:0.1 s。
1.3 原材料纯度核验
对于恩诺沙星、环丙沙星、沙拉沙星、达氟沙星、氧氟沙星、诺氟沙星、洛美沙星7 种国家有证纯度标准物质,直接采用其标示量值和不确定度,对于非有证标准物质培氟沙星,需要对其进行定性和纯度核验。
1.3.1 定性分析
采用高效液相色谱–质谱法和核磁氢谱法对培氟沙星纯品进行定性分析。
(1)高效液相色谱–质谱法。将培氟沙星纯品溶于甲醇中,配制成培氟沙星的质量浓度为1.0 mg/L 的甲醇溶液,采用高效液相色谱–质谱进行全扫描分析。
(2)核磁氢谱法。将培氟沙星纯品溶于氘代甲醇中,采用1H NMR 进行氢谱分析。采样前,调谐和匀场均先采用自动方式,取得初步信号,然后根据化合物信息,再手动调谐、匀场和调节增益。
1.3.2 定量分析
采用高效液相色谱–面积归一化法测定培氟沙星原料的主成分纯度。
1.3.3 杂质定量方法
杂质主要包括水分和无机离子。
水分定量采用卡尔费休水分仪进行测定。准确称量5~10 mg 样品,迅速加入至卡尔费休滴定池内进行滴定,输入称量质量数,读取水分值。为了消除空气和操作过程中水分进入滴定池对测定结果的影响,模拟加样过程,测定实验环境中水分值进行空白扣除。每个样品重复测定5 次,取平均值作为测量结果。
无机离子杂质采用微波消解–电感耦合等离子体质谱法进行测定。称取5 份样品,取样量均为1 g(精确至0.000 1 g),置于微波消解管内,在优化的微波消解条件下进行消解,待消解完全后,取出内罐,冷却至室温,转移至25 mL 容量瓶中,用水定容至标线,混匀备用。同时做试剂空白。
1.3.4 8 种氟喹诺酮类药物互为杂质分析
8 种氟喹诺酮类药物理化性质十分接近,在药物合成或提取过程中,单一组分可能会残留微量同类物质中的其它成分,因此应计算并评估杂质对主成分量值的影响。
1.4 标准物质制备
在(20±2)℃环境温度下,采用重量–容量法配制氟喹诺酮类混合溶液标准物质。分别准确称量8种氟喹诺酮类药物各约50 mg,置于同一500 mL 容量瓶中,用甲醇定容至标线,振荡、混匀,配制成质量浓度均为100 mg/L 的氟喹诺酮类混合溶液标准物质。
将配制好的溶液标准物质在(20±2)℃温度下充分静置,然后分装至350 只2 mL 的洁净棕色安瓿瓶中,分装量为1.2 mL/瓶,编号后置于–18℃冰箱中冷冻保存。
1.5 均匀性检验
从已分装好的溶液标准物质中随机抽取11 瓶样品,按照1.2.1 中的色谱条件进行均匀性检验。以另一瓶独立样品为基准,每瓶样品均重复测定3 次,测定顺序随机,取平均值作为测定结果,用方差分析法统计检验溶液标准物质的均匀性。
1.6 稳定性检验
1.6.1 长期稳定性
长期稳定性一般要求在一定时间内测量点不少于5 个,本研究分别在第0,1,3,6,7 个月随机抽取2 瓶样品,采用与均匀性检验相同的色谱条件,对每瓶样品平行测定3 次,并采用新配制溶液进行单点法浓度定值,采用趋势分析法对监测数据进行评估。
1.6.2 短期稳定性
模拟标准物质运输的实际情况,设定了4,20,50℃3 个不同的温度条件进行短期稳定性考察。分别在第1,3,5,7 d 随机抽取2 瓶样品,采用与长期稳定性相同的方法对每瓶样品平行测定3 次,采用趋势分析法对监测数据进行评估。
2 结果与讨论
2.1 原料定性分析
2.1.1 高效液相色谱–质谱法定性分析
采用高效液相色谱–质谱法,在1.2.1 和1.2.2仪器工作条件下,对培氟沙星纯品进行定性分析。培氟沙星液相色谱–质谱离子峰如图1 所示。

图1 培氟沙星液相色谱–质谱离子峰
由图1 提供的离子碎片信息可知,培氟沙星谱图中有3 个明显的质谱峰,质荷比分别为m/z=334.1,m/z=316.1,m/z=290.3,经过与培氟沙星分子式进行对比分析可知,3 个质谱峰分别属于[M+H]+,[M–OH+H]+,[M–COOH+H]+。此外,谱图中其它信号峰响应均较低。表明分析物为培氟沙星。
2.1.2 核磁氢谱法定性分析
在1.2.3 仪器工作条件下,对培氟沙星进行核磁共振氢谱分析,培氟沙星一维核磁氢谱图见图2。

图2 培氟沙星一维核磁氢谱图
经相位校正和基线校正分析发现,图中化学位移为1.50×10–6的峰为1 位置甲基上氢的特征峰,2.21×10–6的峰为N 相连甲基上氢的特征峰,2.73~3.50×10–6的峰为培氟沙星上含两个N 的六元环上―CH2―位置上氢的特征峰,8.01×10–6的峰为F 旁边CH 位置上氢的特征峰,9.02×10–6的峰是N 旁边CH 位置上氢的特征峰。与检索软件得到的培氟沙星图谱进行对比,两图谱中关键位置的特征峰化学位移均能一一对应,表明分析物为培氟沙星。
2.2 原料定值分析
2.2.1 原料纯度核验
在1.2.1 和1.2.2 仪器工作条件下对培氟沙星进行色谱分析,采用色谱峰面积归一化法测定培氟沙星的纯度。在1.2.4仪器工作条件下,采用微波消解–电感耦合等离子体质谱法测定金属元素含量,用卡尔费休水分仪测定水分含量。图3 为培氟沙星的液相色谱图,培氟沙星纯度核验结果见表3。
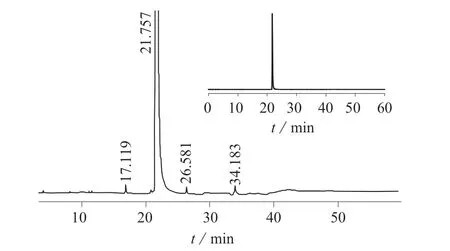
图3 培氟沙星的液相色谱图(总图和局部放大图)

表3 培氟沙星纯度核验结果 %
由表3 可知,实验验证的培氟沙星纯度为92.20%,证书上给定的纯度为92.19%,不确定度为±1%,培氟沙星核验纯度与证书上的标示值基本一致。因此可以采用原纯品证书上的纯度及不确定度。
2.2.2 原料互为杂质分析
将每种氟喹诺酮类药物配制成质量浓度均为100 mg/L 的溶液,采用1.2.1(2)方法进行测定。通过色谱图比较每种药物中杂质的保留时间是否与另外7 种主成分相同,以此判断杂质是否为另外7种主成分,并通过面积归一化得到的百分含量计算其对其它主成分量值的影响,结果见表4。
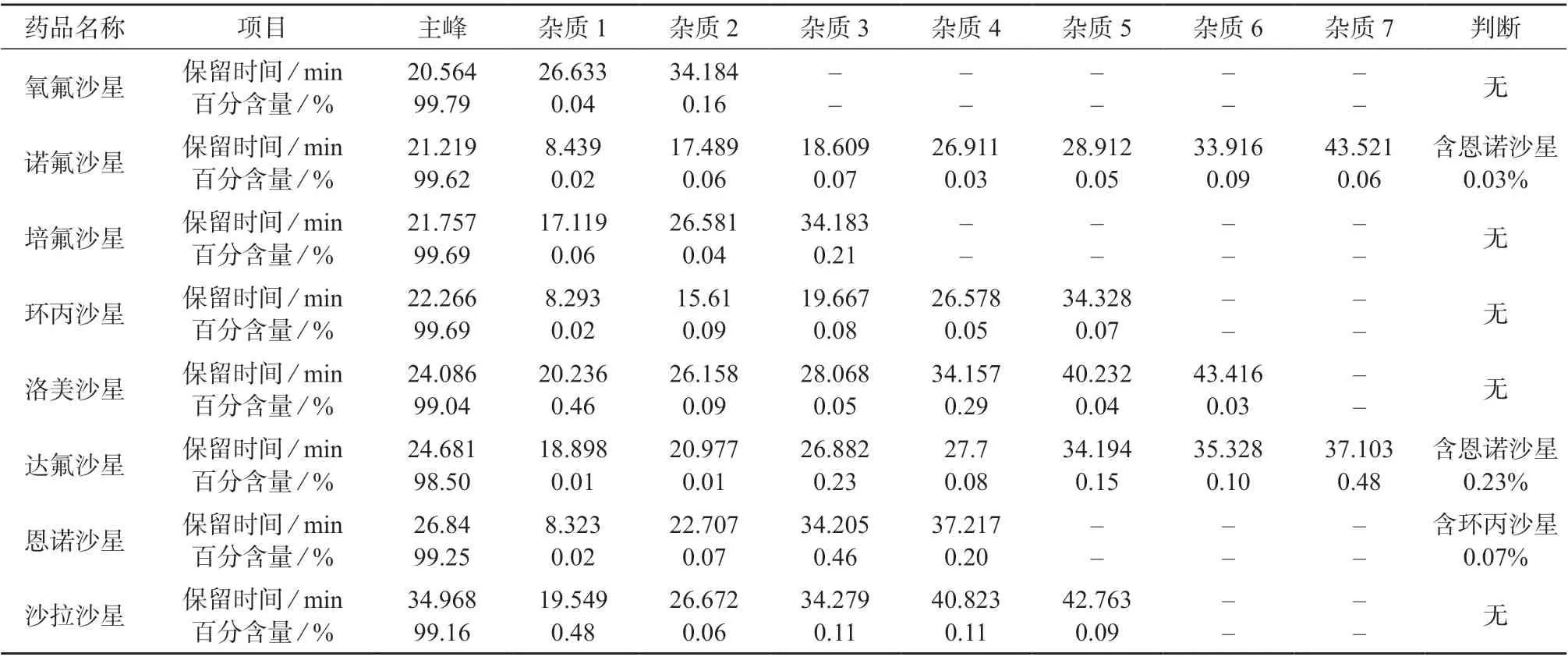
表4 原料互为杂质分析结果
由表4 可知,诺氟沙星和达氟沙星中含有恩诺沙星,含量分别为0.03%和0.23%,因此恩诺沙星的量值应为100.26 mg/L,对于恩诺沙星的量值100 mg/L 几乎没有影响。恩诺沙星中有环丙沙星,但含量仅为0.07%,因此环丙沙星的量值应为100.07 mg/L,对于环丙沙星的量值100 mg/L 几乎没有影响。
2.3 均匀性检验
按照1.5 方法对研制的氟喹诺酮混合溶液标准物质进行均匀性检验,统计结果见表5。

表5 均匀性检验方差分析结果
查表得F0.05(10,22)=2.30,由表5 可知,8 种氟喹诺酮类药物的F值均小于F0.05(10,22),即在95%置信范围内,研制的溶液标准物质中8 种氟喹诺酮类药物的量值在瓶间和瓶内不存在显著性差异,样品均匀性良好。
2.4 稳定性检验
2.4.1 长期稳定性
按照1.6.1 方法进行长期稳定性试验,以检测时间为自变量,以标准物质中8 种氟喹诺酮类药物含量为因变量进行线性回归,计算8 种氟喹诺酮类药物拟合直线的斜率|β1|、截距β0、标准偏差平方s2、斜率不确定度s(β1),结果见表6。
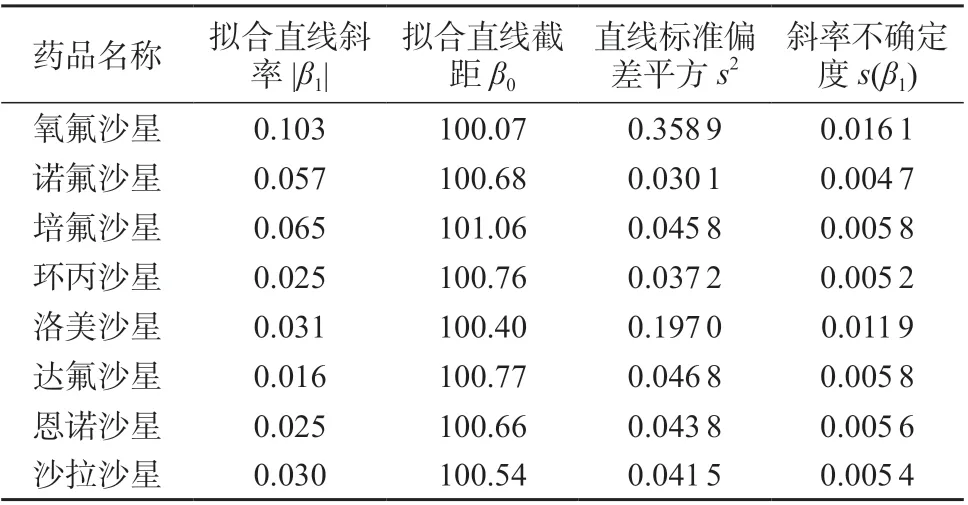
表6 长期稳定性试验结果
查表得t0.95,3=3.18,由表6 可知,8 种氟喹诺酮类药物的|β1|值均小于t0.95,3s(β1),表明直线斜率不显著,研制的标准物质中8 种氟喹诺酮类药物在7个月内的特征量值是稳定的。
2.4.2 短期稳定性
按照1.6.2 方法进行短期稳定性试验,以检测时间为自变量,以标准物质中8 种氟喹诺酮类药物的含量为因变量进行线性回归,计算8 种氟喹诺酮类药物拟合直线的斜率|β1|、截距β0、标准偏差平方s2、斜率不确定度s(β1),结果见表7。

表7 短期稳定性试验结果
由表7 可知,在20℃运输条件下8 种氟喹诺酮类药物的|β1|值均小于t0.95,3s(β1),表明直线斜率不显著,研制的溶液标准物质中8 种氟喹诺酮类药物短期稳定性良好,控制20℃以下的运输条件,7 天内量值稳定。
2.5 不确定评定
氟喹诺酮混合溶液标准物质定值结果的不确定度主要包括溶液配制过程引入的不确定度、均匀性引入的不确定度和稳定性引入的不确定度。
2.5.1 溶液配制过程引入的不确定度
溶液配制过程引入的不确定度主要包括原料纯度、称样质量和定容体积引入的不确定度。
原料纯度引入的不确定度由原料证书获得,结果见表8。
称样质量引入的不确定度主要来自称量的变动性和天平校正。可以通过天平的检定证书获得,天平重复性检定结果分别为0.08 mg 和0.03 mg,假设正态分布,则换算成标准偏差后将两项合成得称样质量引入的标准不确定度,结果见表8。

将原料纯度、称样质量和定容体积引入的不确定度合成得溶液配制过程引入的相对标准不确定度,结果见表8。

表8 溶液配制过程引入的相对标准不确定度 %
2.5.2 均匀性引入的不确定度
均匀性引入的不确定度分量主要来源于标准物质制备过程、分装以及实验分析间的偏差。标准物质的不均匀性以独立测量的平行试验来表示,即以瓶间与瓶内均匀性检验结果的相对标准偏差来表示。
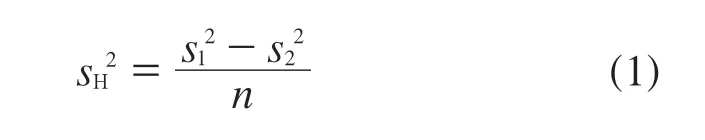

式中:sH——瓶间标准偏差;
——组间方差;
——组内方差;
n——组内测量次数;
υ2——组内自由度。
在这种情况下,sH等同于瓶间不均匀性引入的不确定度分量ubb,即公式ubb=sH。
将表5 中数据代入上式,计算得均匀性引入的不确定度,结果见表9。
2.5.2 稳定性引入的不确定度
根据ISO 导则35[19],甲醇中8 种氟喹诺酮类药物混合溶液标准物质稳定性引入的不确定度u1ts由式(3)计算:

式中:s(β1)——稳定性检测数据拟合直线斜率的相 对标准不确定度;
t——给定的保存期限。
分别将表6 和表7 中数据代入式(3),计算得长期、短期稳定性引入的不确定度,结果见表9。
2.5.4 总不确定度
将上述各不确定度分量合成得氟喹诺酮混合溶液标准物质总不确定度和扩展不确定度(k=2,取整数),结果见表9
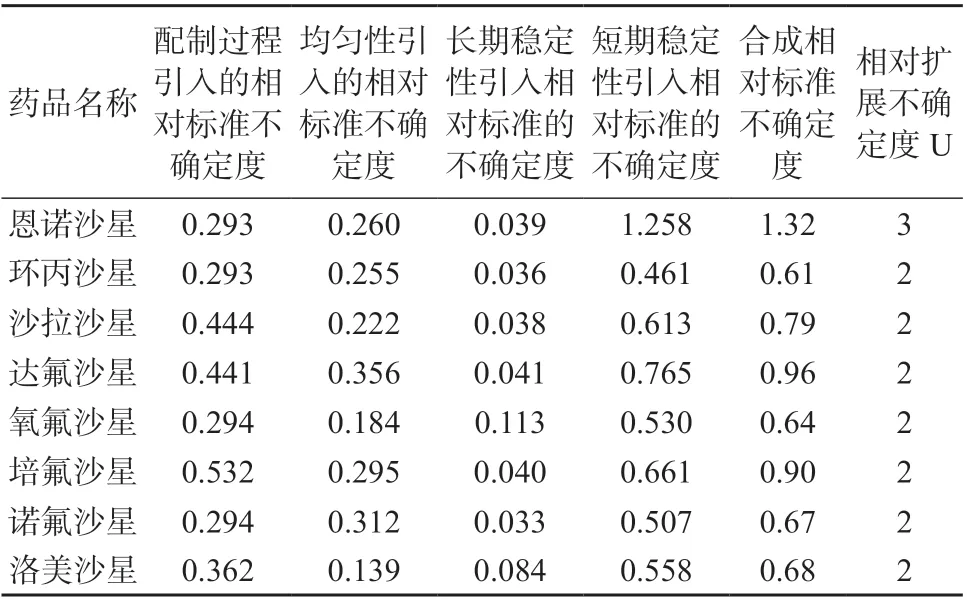
表9 溶液标准物质不确定评估结果 %
3 结语
以恩诺沙星、环丙沙星、沙拉沙星、达氟沙星、氧氟沙星、诺氟沙星、洛美沙星7 种国家有证标准物质和经纯度核验后的培氟沙星纯品为原料,采用重量–容量法制备了甲醇中8 种氟喹诺酮药物混合溶液标准物质。考察了8 种氟喹诺酮互为杂质情况,利用高效液相色谱法进行了均匀性检验与稳定性检验,对配制过程中产生的不确定度进行了系统的分析和评定。该混合溶液标准物质已被批准为国家二级标准物质,编号为GBW(E) 083598,为农产品质量安全监测与风险评估中氟喹诺酮类药物残留测定提供了溯源保障。
